

 下载:
下载:

-
高砷地下水在全球范围内广泛分布,对人类的饮用水安全有严重威胁[1, 2]. 地下水中砷(As)主要有As(Ⅲ)和As(Ⅴ)两种形式,二者以溶解态存在,具有较强的迁移性. 研究表明,地下水中砷的迁移能力与沉积物中铁(Fe)含量存在显著相关性[3, 4]. 铁元素主要存在于铁氧化物和含铁黏土中[5 − 7],大量实验数据表明,微生物驱动的铁氧化物的还原溶解是高砷地下水形成的主要原因[7- 8]. 与铁氧化物不同,含铁黏土矿物属于含水层状硅酸盐矿物[9],铁元素存在于其层状结构单元骨架中,因此,Fe(Ⅲ)的还原不会造成含铁黏土矿物的溶解[10]. 此外,含铁黏土矿物具有高比表面能和强吸附性,常存在于沉积物中,能够作为砷或其他污染物吸附/释放的汇/源[11],对地下水中污染物的迁移转化有重要的影响[12 − 13]. 常见的含铁黏土矿物有高岭石、蒙脱石、绿脱石等,其中绿脱石 (nontronite NAu-2,以下简称为NAu-2)是一种富铁的黏土矿物,其铁含量可高达4.1 mmol·g−1 Fe [14],对砷的迁移能力具有显著影响[11, 15],是研究黏土矿物与砷迁移转化过程的理想材料. 然而,目前黏土矿物表面吸附态砷的提取方法研究较少,限制了对砷迁移转化过程的研究.
目前土壤中砷的提取主要依赖于化学方法,如利用浓盐酸直接提取[16]、强碱超声提取[17]、磷酸盐和氨盐分步提取[18]等,均可提取土壤中的总砷. 然而,Wang等[19]发现,在提取粉煤灰中砷的过程中,As(Ⅲ)被氧化为As(Ⅴ),这一现象引起了人们对提取过程中可能发生的砷价态变化的关注. Pantsarkallio等[20]在提取含As(Ⅴ)的土壤样品时,未观察到价态的变化;而提取含As(Ⅲ)的土壤样品时,大量As(Ⅲ)被氧化,导致As(Ⅲ)回收率最低仅有20%. 同样,Thomas等[21]在提取不同来源的含砷样品中也观察到了类似的现象,在氧化环境土壤中As(Ⅴ)的价态不受影响,在还原环境的河流沉积物提取中则发生As(Ⅲ)的氧化反应. 这些研究表明,提取过程对As(Ⅴ)没有影响,而As(Ⅲ)的氧化现象则普遍存在.
为了解决这一问题,本研究采用提取剂中加入弱还原性的“还原保护剂”的思路,抑制提取过程的氧化反应发生,将黏土矿物表面吸附态砷以原价态提取至溶液中,为黏土矿物与砷迁移转化机制研究奠定基础. 本文建立了一种能够高效且分价态的同步提取含铁黏土矿物表面砷的方法. 针对氧化态Fe(Ⅲ)-NAu-2和还原态Fe(Ⅱ)-NAu-2表面吸附的As(Ⅲ)和As(Ⅴ),以传统的酸提取法为基础,结合两种体系中铁、砷和提取剂间的氧化还原作用机制,筛选出最佳提取剂种类、提取剂浓度、提取时间,通过添加有效的还原保护剂,实现含铁黏土矿物表面吸附的As(Ⅲ)和As(Ⅴ)的同步提取,为更深入地研究含铁黏土矿物富集的地下水环境中砷的迁移和转化过程提供基础支持.
-
磷酸(H3PO4)、硫酸(H2SO4)、盐酸(HCl)、氢氧化钠(NaOH)、亚砷酸钠(NaAsO2)和砷酸钠(Na2HAsO4·7H2O)购于中国国药集团化学试剂有限公司. 1,4-哌嗪二乙磺酸(以下简称为PIPES)、磷酸二氢钾(KH2PO4)、磷酸氢二钠(Na2HPO4)、柠檬酸钠(C6H5Na3O7)、乳酸钠(C3H5NaO3)、盐酸羟胺(NH2OH·HCl)和硼氢化钾(KBH4)购自上海阿拉丁生化科技股份有限公司. 药品均为分析纯或优级纯. 实验用水均为超纯水(18.2 MΩ·cm).
微生物Shewanella oneidensis MR-1(以下简称为MR-1) 购买于美国标准微生物库ATCC,MR-1培养基为不含葡萄糖的胰蛋白胨大豆肉汤[22](以下简称为TSB-D培养基),培养基中包含:胰蛋白胨(BactoTM Tryptone)、大豆蛋白胨(BactoTM Soytone)、磷酸氢二钾(K2HPO4)、氯化钠(NaCl),上述药品均购自上海阿拉丁生化科技股份有限公司. 含铁黏土矿物NAu-2购买于美国黏土矿物协会,化学式为M+0.72[Si7.55Al0.16Fe0.29][Al0.34Fe3.54Mg0.05]O20(OH)4,其中M+为吸附的阳离子. 将NAu-2研磨分散至0.5 mol·L−1 NaCl 溶液中,进行搅拌、超声等操作,分离出粒径为0.5—2.0 μm的NAu-2,超纯水反复洗涤离心,直到上清液中不能检测到Cl-,60 ℃下干燥.
-
As(Ⅴ)储备液由Na2HAsO4·7H2O配制,As(Ⅲ)储备液由NaAsO2溶于无氧超纯水配制,储备液砷浓度均为1 g·L−1,避光密封保存于4 ℃恒温冰箱中. 氢氧化钠固体溶解于超纯水中制备1 mol·L−1 NaOH溶液. 10 mmol·L−1 PIPES缓冲液由PIPES溶于超纯水并调节pH=7.0制得. NAu-2储备液(20 g·L−1)由PIPES缓冲液配制备用. 乳酸钠溶于10 mmol·L−1 PIPES得到50 mmol·L−1乳酸钠储备液. 上述储备液均经过灭菌处理.
配制TSB-D培养基并调节pH为7.2,121℃高压灭菌15 min,于超净台中接种MR-1菌种,培养至对数期,无氧PIPES溶液(经高纯氮气曝气脱氧)洗涤离心3次,最后取重悬液稀释,进行浓度测定备用. 所有实验均在室温 (25 ℃) 下进行.
-
在铝箔包覆的密封西林瓶中构建20 mL反应体系. PIPES缓冲液模拟地下水中性环境,体系中MR-1浓度为1×108 cells·mL−1,NAu-2的浓度为2 g·L−1,电子供体乳酸钠浓度为5 mmol·L−1,置于转速为150 r·min−1的摇床上振荡. 生物还原过程持续时间约60 h,灭菌得到还原态Fe(Ⅱ)-NAu-2备用. 整个实验过程均在厌氧手套箱中进行.
-
将As(Ⅲ)和As(Ⅴ)分别加入“1.3”节中的还原态Fe(II)-NAu-2体系中,构建还原态Fe(Ⅱ)-NAu-2-As(Ⅲ)和还原态Fe(Ⅱ)-NAu-2-As(Ⅴ)体系,其中As(Ⅲ)和As(Ⅴ)的初始浓度均为200 μg·L−1. 加入的As(Ⅲ)和As(Ⅴ)对体系其他物质浓度造成的影响可忽略不计. 在铝箔包覆的密封西林瓶中构建20 mL氧化态Fe(Ⅲ)-NAu-2-As反应体系,PIPES作为缓冲液,体系中NAu-2浓度为2 g·L−1,乳酸钠溶液为5 mmol·L−1,As初始浓度为200 μg·L−1. 整个实验过程均在厌氧手套箱中进行. 待溶液中的砷浓度稳定,则认定该体系已达到吸附平衡,本研究中吸附反应时间100 h.
-
以“1.4”节中构建的吸附平衡体系作为初始提取状态,从西林瓶移取0.5 mL NAu-2-As样品注入离心管中,加入提取剂后,置于转速为150 r·min−1的摇床上振荡60 min,高速离心机离心(
10000 r·min−1)10 min,快速吸取上清液,经0.22 μm滤膜过滤后,存放于棕色样品瓶中,获得提取后的砷溶液,进行浓度测定. 提取效率计算公式如下:式中:
$ {C}_{a} $ 为加入实验体系中的总As浓度,单位μg·L−1,本实验中$ {C}_{\mathrm{a}} $ 为200 μg·L−1;$ {C}_{0} $ 为NAu-2-As吸附平衡体系的溶液中As浓度,单位μg·L−1;$ {C}_{t} $ 为提取剂提取吸附态砷后溶液中的As浓度,单位μg·L−1;$ {\phi }_{\mathrm{提}} $ 为提取效率. -
本实验采用对砷具有提取效果的磷酸二氢钾(10 mmol·L−1 KH2PO4)、盐酸(10 mmol·L−1 HCl)、硫酸(10 mmol·L−1 H2SO4)、磷酸(7.5% H3PO4(V/V))、磷酸二氢钾-氢氧化钠(10 mmol·L−1 KH2PO4-0.5 mol·L−1 NaOH)5种化学试剂,分别提取Fe(Ⅲ)-NAu-2-As(Ⅲ)平衡体系和Fe(Ⅲ)-NAu-2-As(Ⅴ)平衡体系中黏土矿物表面的吸附态砷,选取效果较好的提取剂.
-
本部分实验考察体积浓度分别为7.5%、4%、2%、1%以及0.5%的磷酸提取剂对吸附在Fe(Ⅲ)-NAu-2表面的As(Ⅲ)和As(Ⅴ)的提取效率(本文中磷酸浓度均指体积浓度),确定提取剂最佳浓度.
-
本实验将提取时间设置为5、10、15、30、60 min,分析氧化态Fe(III)-NAu-2表面砷的提取效率随时间的变化情况,构建提取效率与提取时间之间的关系.
-
考察在不同初始As(Ⅲ)和As(Ⅴ)浓度(50、100、200、500、
1000 μg·L−1)下磷酸的提取效率,分析砷初始浓度对Fe(Ⅲ)-NAu-2表面吸附态砷提取效率的影响,同时确定磷酸提取适用的砷浓度提取范围. -
在Fe(Ⅱ)-NAu-2-As(Ⅲ)和Fe(Ⅱ)-NAu-2-As(Ⅴ)体系中,待吸附反应达到平衡后进行提取方法研究. 为避免磷酸提取过程中发生氧化还原反应造成提取砷价态发生变化,在对还原态Fe(Ⅱ)-NAu-2-As体系中砷提取时加入浓度为100 mmol·L−1的还原剂,分别考察以磷酸-抗坏血酸、磷酸-柠檬酸钠、磷酸-盐酸羟胺等强酸-弱还原剂组合作为提取剂,构建能够使砷以原价态脱附于还原态Fe(Ⅱ)-NAu-2表面的高效提取方法.
-
不同价态砷浓度由高效液相色谱(HPLC,LC20A,岛津,日本)与原子荧光光谱(AFS-2202E,海光,中国)联用测定[23]. 液相所用的分离柱为PRP-X100阴离子交换柱(4.1 mm×250 mm),磷酸二氢钾(45 mmol·L−1)和磷酸氢二钠(5 mmol·L−1)为流动相,流速为1 mL·min−1;经液相分离后的As(Ⅲ)和As(Ⅴ)在5%(V/V)盐酸和2%(W/V)的硼氢化钾共同作用下,经原子荧光光谱仪原子化后在砷灯(海光,中国)的照射下形成电信号,分别进行浓度检测. 载气和保护气均为高纯氩气(99.99%),载气流速为500 mL·min−1.
-
氧化态Fe(Ⅲ)-NAu-2和还原态Fe(II)-NAu-2对As(Ⅲ)和As(Ⅴ)的吸附平衡结果如图1所示. 氧化态和还原态的NAu-2对As(Ⅲ)和As(Ⅴ)均具有吸附能力,且对As(Ⅴ)的吸附能力强于As(Ⅲ),主要是由于As(Ⅲ)和As(Ⅴ)在中性缓冲溶液中形态不同,As(Ⅲ)主要以不带电的H3AsO3分子存在,通过羟基配位体交换的方式吸附于矿物表面[24 − 25],而As(Ⅴ)以HAsO42-阴离子形式存在,带负电的As(Ⅴ)通过静电引力与阳离子桥接的作用吸附在黏土矿物表面,因此NAu-2对As(Ⅴ)有更强的吸附能力[26].
在Fe(Ⅲ)-NAu-2-As(Ⅲ)和 Fe(Ⅲ)-NAu-2-As(Ⅴ)体系达到吸附平衡时,As(Ⅲ)和As(Ⅴ)的吸附浓度分别为66.8 μg·L−1和161.5 μg·L−1,Fe(Ⅲ)-NAu-2对As(Ⅲ)和As(Ⅴ)的吸附率分别为33.4%和80.8%. 而NAu-2被微生物还原后,还原态Fe(Ⅱ)-NAu-2对As(Ⅲ)和As(Ⅴ)的吸附率分别为14.3%和21.4%,说明NAu-2的生物还原会导致其对砷的吸附能力下降. 以吸附平衡时的体系作为黏土矿物表面吸附态砷提取的初始状态,分别针对Fe(Ⅲ)-NAu-2-As和Fe(Ⅱ)-NAu-2-As体系中吸附态砷进行提取实验,构建吸附态砷的高效提取方法.
-
不同提取剂对黏土矿物表面吸附态砷的提取效率不同. 本实验选取五种常用的砷提取剂,按照“1.5”节中所述的吸附态砷的提取方法,以磷酸二氢钾(KH2PO4)[27]、盐酸(HCl)[28]、硫酸(H2SO4)、磷酸(H3PO4)[27]、磷酸二氢钾-氢氧化钾(KH2PO4-NaOH)[17]对氧化态Fe(Ⅲ)-NAu-2表面As(Ⅲ)和As(Ⅴ)的提取效率如图2所示. 提取结果表明,磷酸、硫酸和盐酸等3种酸性提取剂对吸附态As(Ⅴ)提取效果较好,其中磷酸提取后溶液中As(Ⅴ)浓度为185.6 μg·L−1,对黏土矿物表面吸附态As(Ⅴ)的提取效率最高,可达91.1%,硫酸和盐酸提取效果次之,提取率分别为84.5%和81.0%;而磷酸二氢钾-氢氧化钠、磷酸二氢钾两种盐类提取剂提取后溶液中As(Ⅴ)浓度仅有152.8 μg·L−1和164.1 μg·L−1,提取效率均低于80%,效果较差. 上述提取剂在提取过程中均未发生As(Ⅴ)价态的转化.
图2中展示了5种提取剂对氧化态Fe(Ⅲ)-NAu-2-As(Ⅲ)体系中吸附态As(Ⅲ)的提取效率. 磷酸和硫酸对吸附态As(Ⅲ)的提取效率分别达到81.3%和84.7%,盐酸提取后溶解态As(Ⅲ)浓度为163.9 μg·L−1,提取效率为72.9%,相较于硫酸提取效率降低了11.8%,磷酸二氢钾-氢氧化钠和磷酸二氢钾对As(Ⅲ)的提取效率均小于60%,远小于磷酸和硫酸的提取效率. 根据上述结果,磷酸和硫酸对As(Ⅲ)和As(Ⅴ)的提取效率均高于81%,且提取过程未发生砷价态的转化,可作为Fe(Ⅲ)-NAu-2-As体系中吸附态As(Ⅲ)和As(Ⅴ)的高效提取剂.
磷酸和硫酸属于强酸,黏土矿物在强酸性条件下易发生质子化反应[29]. 质子化反应会增加黏土矿物表面的正电荷,降低对As(Ⅴ)的吸附能力[30],同时大量H+的引入使黏土矿物边面裸露的羟基脱除,减少了As(Ⅲ)的可能吸附位点,最终导致吸附在黏土矿物表面上As(Ⅲ)和As(Ⅴ)脱附[29, 31]. 研究发现,磷酸根与砷酸根具有相似的化学行为,能够特异性吸附在土壤表面,当土壤溶液中磷酸根浓度增加时,砷酸根的吸附速率和吸附量显著降低[32],证明磷酸的加入会使磷酸根与吸附态砷竞争吸附位点[33- 34]. 此外,磷酸根在层状硅酸盐矿物表面的亲和力强于砷酸根[34],有利于黏土矿物表面砷的脱附. 因此,以磷酸作为提取剂,可通过质子化和竞争吸附两种途径促进黏土矿物表面的吸附态砷脱附,更易提取黏土表面的吸附态砷,因此选用磷酸作为后续实验提取剂.
-
根据以上实验可知,磷酸对黏土矿物表面吸附态砷具有最佳的提取效果,此时磷酸浓度为7.5%(V/V),磷酸浓度较高,在原子荧光测定As(Ⅲ)和As(Ⅴ)的信号峰出现明显的拖尾现象,严重影响As(Ⅲ)和As(Ⅴ)浓度测定的准确性. 因此本部分研究中分析了降低磷酸体积浓度(0.5%—7.5%)对Fe(Ⅲ)-NAu-2表面吸附的As(Ⅲ)和As(Ⅴ)提取效率的影响(如图3). 结果表明,磷酸浓度的降低对As(Ⅴ)的提取效率影响较小,体积浓度为0.5%—7.5%的磷酸均可实现对As(Ⅴ)的高效提取,提取率范围为90.0%—92.5%,当磷酸浓度为1%和2%时,对As(Ⅴ)提取率分别可达92.5%和92.0%. 然而,磷酸浓度变化会显著影响As(Ⅲ)的提取效率,浓度为1%的磷酸对As(Ⅲ)的提取效果最好,提取率为85.0%,当磷酸浓度为2%时,对As(Ⅲ)提取率仅为79.4%,其余浓度的磷酸溶液对As(Ⅲ)提取效率均低于82%. 因此,体积浓度为1%的磷酸是提取吸附态砷的最佳浓度.
-
提取剂与样品悬浊液的接触时间是影响提取效率的关键. 研究发现,土壤中砷的提取时间一般设置为60 min[28, 35],提取时间过长不但影响实验的效率,还可能影响砷的价态. 磷酸提取时间缩短对As(Ⅲ)和As(Ⅴ)提取效率的影响如图4所示. 结果表明,提取时间在5—60 min的范围内,As(Ⅲ)和As(Ⅴ)提取效率分别为82.1—88.0%和92.6%—95.2%. 磷酸提取下吸附态砷的释放是一个快速的过程,向体系中加入磷酸,大量的H+的进入体系,使化学吸附的带负电的砷酸根阴离子占比大量减少,同时会造成通过静电吸附的亚砷酸分子快速释放[36, 37]. 结合实验结果,在提取时间为10 min时,As(Ⅲ)和As(Ⅴ)的提取率均可同时达到最高,分别为88.0%和95.2%,因此将最佳提取时间均设定为10 min.
-
根据以上实验,采用体积浓度为1%的磷酸对不同初始浓度的As(Ⅲ)和As(Ⅴ)进行提取,10 min后提取效率如图5所示. 结果表明,初始砷浓度为200 μg·L−1,As(Ⅲ)和As(Ⅴ)的提取效率可达88.0%和95.2%;初始砷浓度为500 μg·L−1时,As(Ⅲ)和As(Ⅴ)的提取效率分别为83.8%和95.9%;初始砷浓度为
1000 μg·L−1时,As(Ⅲ)和As(Ⅴ)的提取效率分别为81.6%和94.6%. 当初始砷浓度为50 μg·L−1和100 μg·L−1时,磷酸提取对As(Ⅴ)的提取效率均高于91%,但是对As(Ⅲ)的提取效率仅有78.9%和76.7%. 因此,初始砷浓度由200 μg·L−1逐渐提高到1000 μg·L−1,不会影响磷酸As(Ⅴ)的提取效率,As(Ⅲ)的提取率虽然略有降低,但均高于80%;在50—100 μg·L−1的砷浓度范围内,磷酸对As(Ⅴ)可以保持较高提取率,而磷酸对As(Ⅲ)的提取能力显著下降. 提取过程并未发生As的价态转化. 该实验也表明此方法适用于200—1000 μg·L−1的高砷浓度范围内氧化态Fe(Ⅲ)-NAu-2表面吸附态砷的提取.综上所述,通过实验条件的对比与筛选,在Fe(Ⅲ)-NAu-2-As体系中,用体积浓度为1%的磷酸对NAu-2表面吸附的As(Ⅴ)和As(Ⅲ)提取10min后,As(Ⅴ)和As(Ⅲ)的提取率均高于80%,且提取过程中As(Ⅴ)和As(Ⅲ)价态无变化,可实现对Fe(Ⅲ)-NAu-2表面吸附态砷的高效提取.
-
直接采用“2.2”中的磷酸提取法对还原态Fe(Ⅱ)-NAu-2表面吸附的As(Ⅴ)和As(Ⅲ)进行提取,结果如图6所示. 其中空白对照组为还原态Fe(Ⅱ)-NAu-2与As(Ⅲ)和As(Ⅴ)的吸附平衡态,即未提取状态下溶液中的砷浓度. 在Fe(Ⅱ)-NAu-2-As(Ⅴ)体系中(图6a),相对于空白对照组,磷酸提取后As(Ⅴ)浓度由157.2 μg·L−1增高到192.8 μg·L−1,提取率为83.1%,表明在磷酸作用下可高效提取Fe(Ⅱ)-NAu-2表面吸附的As(Ⅴ),且磷酸提取后在溶液几乎未检测到As(Ⅲ),说明提取过程中As(Ⅴ)价态并未发生变化.
在Fe(Ⅱ)-NAu-2-As(Ⅲ)体系中,溶液中空白对照组中部分As(Ⅲ)被氧化为As(Ⅴ),As(Ⅴ)浓度为12.4 μg·L−1,这是由于NAu-2中部分Fe(Ⅲ)被生物还原为Fe(II)后,将形成具有强氧化性的Fe(Ⅱ)-O-Fe(Ⅲ)键,该键能够将As(Ⅲ)氧化为As(Ⅴ)[5, 38]. 用磷酸直接进行As(Ⅲ)提取后,发现溶液中As(Ⅴ)浓度增加到17.9 μg·L−1,增幅高达44.4%. 以上结果表明,直接采用磷酸可对Fe(Ⅱ)-NAu-2表面吸附的As(Ⅴ)进行高效提取,但在Fe(II)-NAu-2-As(Ⅲ)体系中,磷酸的提取过程会导致As(Ⅲ)的氧化.
-
根据文献资料,本研究选取抗坏血酸、柠檬酸钠和盐酸羟胺三种物质作为还原保护剂[39 − 40],将还原保护剂(100 mmol·L−1)与磷酸(1%)同时添加到Fe(II)-NAu-2-As(Ⅴ)体系中进行As(Ⅴ)提取(图6a). 以磷酸-抗坏血酸与磷酸-柠檬酸钠作为提取剂,溶液中As(Ⅴ)的浓度分别为193.0 μg·L−1和192.0 μg·L−1,提取率为83.6%和81.3%. 以磷酸-盐酸羟胺作为提取剂,溶液中As(Ⅴ)的浓度为194.8 μg·L−1,As(Ⅴ)的提取率最高,可达87.8%. 在提取过程中未检测到As(Ⅴ)价态的转化. 结果表明磷酸-盐酸羟胺优于磷酸-抗坏血酸与磷酸-柠檬酸钠的提取效果,因此磷酸-盐酸羟胺可作为还原态Fe(Ⅱ)-NAu-2-As(Ⅴ)体系的高效提取剂.
添加弱还原性保护剂后,Fe(Ⅱ)-NAu-2-As(Ⅲ)体系中As(Ⅲ)的提取效率如图6b. 结果表明,与空白组相比,以磷酸-抗坏血酸为提取剂,溶液中As(Ⅲ)浓度为175.3 μg·L−1,As(Ⅴ)浓度由12.4 μg·L−1升高至20.5 μg·L−1,溶液中As(Ⅴ)浓度的增长率高达65.3%,说明抗坏血酸并未抑制提取过程中砷的价态转化. 以磷酸-柠檬酸钠作为提取剂,溶液中As(Ⅲ)浓度为172.6 μg·L−1,As(Ⅴ)浓度升高至21.8 μg·L−1,As(Ⅴ)浓度的大幅度升高同样表明在提取过程中As(Ⅲ)的氧化并未得到有效抑制. 以磷酸-盐酸羟胺作为提取剂,溶液中As(Ⅲ)浓度由157.2 μg·L−1升高至184.9 μg·L−1,As(Ⅴ)浓度为12.5 μg·L−1,总砷的提取效率高达93.9%. 以上数据表明以磷酸-盐酸羟胺作为提取剂能够高效提取As(Ⅲ),且能有效避免在Fe(II)-NAu-2表面吸附As(Ⅲ)提取过程中的氧化,因此选取盐酸羟胺作为磷酸提取时的还原保护剂.
根据以上实验结果,针对还原态Fe(Ⅱ)-NAu-2-As体系在提取过程中会发生价态转化的难题,选择磷酸-盐酸羟胺作为提取剂,既能够将吸附态As(Ⅲ)和As(Ⅴ)分价态提取至溶液中,也能实现对吸附态砷的高效提取.
-
本实验针对氧化态Fe(Ⅲ)-NAu-2和还原态Fe(Ⅱ)-NAu-2表面吸附态As(Ⅲ)和As(Ⅴ)同步提取方法进行研究,得出以下结论:
1)在氧化态Fe(Ⅲ)-NAu-2-As体系中,可用磷酸直接进行Fe(Ⅲ)-NAu-2表面吸附态As(Ⅲ)和As(Ⅴ)的提取,采用体积浓度为1%的磷酸溶液作为提取剂,提取时间10 min,表面吸附态As(Ⅲ)和As(Ⅴ)的提取率分别达到88.0%和95.2%,该方法适用于砷浓度范围为200—
1000 μg·L−1的黏土矿物表面吸附态砷提取,且提取过程中As(Ⅴ)和As(Ⅲ)均无价态转化.2)在还原态Fe(Ⅱ)-NAu-2-As体系中,通过添加100 mmol·L−1盐酸羟胺抑制提取过程中发生的氧化反应,实现Fe(Ⅱ)-NAu-2表面吸附的As(Ⅲ)和As(Ⅴ)的原价态提取,砷提取效率分别为93.9%和87.8%.
3)本文构建了黏土矿物表面As(Ⅲ)和As(Ⅴ)同步提取的方法,有效的解决了黏土矿物表面吸附态砷提取过程中的氧化问题. 以磷酸和磷酸-盐酸羟胺分别作为氧化态Fe(Ⅲ)-NAu-2和还原态Fe(Ⅱ)-NAu-2表面吸附As(Ⅴ)和As(Ⅲ)的提取剂,能够在砷价态不变的前提下实现高效提取. 同时,该方法可推广至其他含有诱导砷价态转化的土壤沉积物样品中表面吸附态As(Ⅲ)和As(Ⅴ)的同步提取,对深入研究环境中砷的迁移转化过程具有重要意义.
含铁黏土矿物表面吸附态As(Ⅲ)和As(Ⅴ)的同步提取方法
Study on the simultaneous extraction method of As (Ⅲ) and As (Ⅴ) adsorbed on the surface of iron-bearing clay minerals
-
摘要: 砷(As)是一种典型的有毒污染物,其在地下环境中的迁移转化与铁(Fe)的氧化还原过程密切相关. 含铁黏土矿物广泛分布于沉积物中,不同于铁氧化物,铁元素以结构态赋存于黏土矿物结构骨架中,环境条件变化不会导致结构态铁的还原性溶解. 然而,含铁黏土矿物中结构态铁的氧化还原不仅影响砷的迁移能力,而且对砷的价态转化有着重要影响. 黏土矿物表面吸附态砷的分价态高效提取是研究黏土矿物与砷迁移转化研究的前提,但目前尚无完善的分价态同步提取黏土矿物表面吸附态砷的方法. 本文以含铁黏土矿物NAu-2为例,建立了氧化态Fe(Ⅲ)-NAu-2和还原态Fe(Ⅱ)-NAu-2表面吸附态As(Ⅲ)与As(Ⅴ)的分价态提取分析方法. 结果表明,采用体积浓度为1%的磷酸作为提取剂,提取10 min,对Fe(Ⅲ)-NAu-2-As(Ⅲ)和Fe(Ⅲ)-NAu-2-As(Ⅴ)体系中砷提取率分别达到88.0%和95.2%. 以体积浓度为1%的磷酸作为提取剂,100 mmol·L-1盐酸羟胺为还原保护剂,提取时间为10 min,Fe(Ⅱ)-NAu-2-As(Ⅲ)和Fe(Ⅱ)-NAu-2-As(Ⅴ)体系中砷的提取率分别可达93.9%和87.8%. 本研究解决了含铁黏土矿物吸附态砷提取至溶液过程中的价态变化难题,实现了As(Ⅲ)和As(Ⅴ)在黏土矿物表面的高效同步提取,对进一步研究自然环境中含铁黏土矿物对砷迁移转化过程的影响机制具有重要意义.Abstract: Arsenic (As) is a typical toxic pollutant, and its migration and transformation in the subsurface environment are closely associated with the redox processes of iron (Fe). Iron-bearing clay minerals are widely distributed in sediments. Unlike iron oxides, iron element exists as structural iron within the mineral framework, and changes in environmental conditions can’t lead to the reductive dissolution of structural iron. However, the redox reactions of structural iron in iron-bearing clay minerals not only affects the mobility of arsenic, but also has a significant impact on the valence state transformation of arsenic. The efficient extraction of arsenic in different valence states adsorbed on the surface of clay minerals is a prerequisite for studying the migration and transformation of arsenic with clay minerals. Nevertheless, there is currently no established method for the simultaneous extraction of different valence states of arsenic adsorbed on the surface of clay minerals. In this study, using nontronite NAu-2 as an example, a method for extracting the valence states of As(Ⅲ) and As(Ⅴ) absorbed on the surface of oxidized Fe(Ⅲ)-NAu-2 and reduced Fe(Ⅱ)-NAu-2 was established. The results indicate that using 1% (V/V) of phosphoric acid as the extractant for 10 minutes, the extraction rates of arsenic in the Fe(Ⅲ)-NAu-2-As(Ⅲ) system and Fe(Ⅲ)-NAu-2- As(Ⅴ) system reach 88.0% and 95.2%, respectively. With 1% (V/V) of phosphoric acid as the extractant and 100 mmol·L-1 hydroxylamine hydrochloride as the reducing protectant, the extraction rates of arsenic in the Fe(Ⅱ)-NAu-2-As(Ⅲ) system and Fe(Ⅱ)-NAu-2-As(Ⅴ) system can reach 93.9% and 87.8% respectively, with an extraction time of 10 minutes. This study addresses the difficulty of valence state changes during the extraction of adsorbed arsenic in iron-bearing clay minerals, achieving the efficient simultaneous extraction of As(Ⅲ) and As(Ⅴ) on the surface of clay minerals. This has significant implications for further studying the impact mechanism of iron-bearing clay minerals on the migration and transformation of arsenic in the natural environment.
-
Key words:
- arsenic speciation /
- nontronite /
- phosphoric acid extraction /
- reducing protectant.
-
-
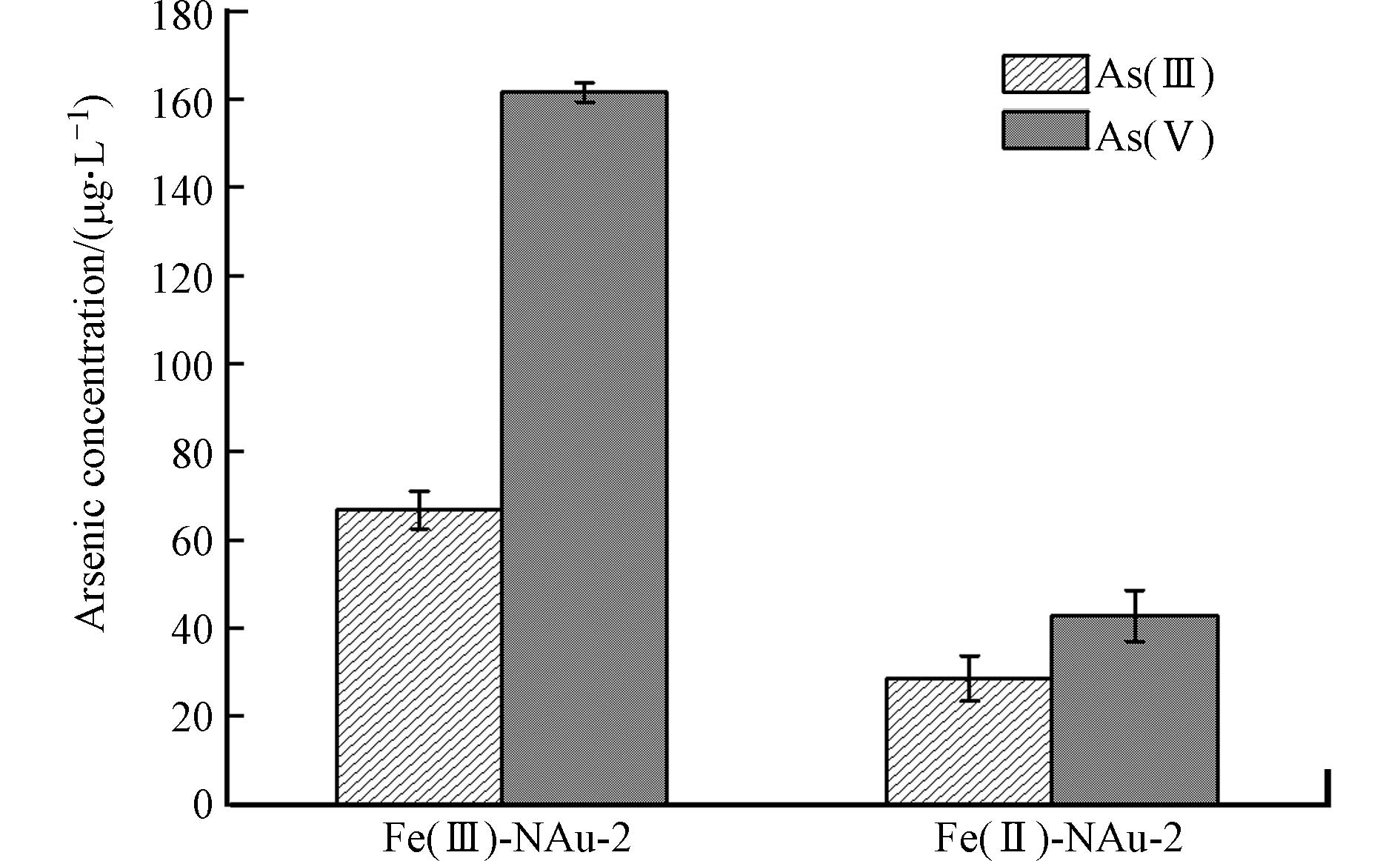
图 1 氧化态Fe(Ⅲ)-NAu-2和还原态Fe(Ⅱ)-NAu-2对As(Ⅲ)和As(Ⅴ)的吸附浓度
Figure 1. Adsorption concentration of As (Ⅲ) and As (Ⅴ) by oxidized Fe (Ⅲ)-NAu-2 and reduced Fe (Ⅱ)-NAu-2
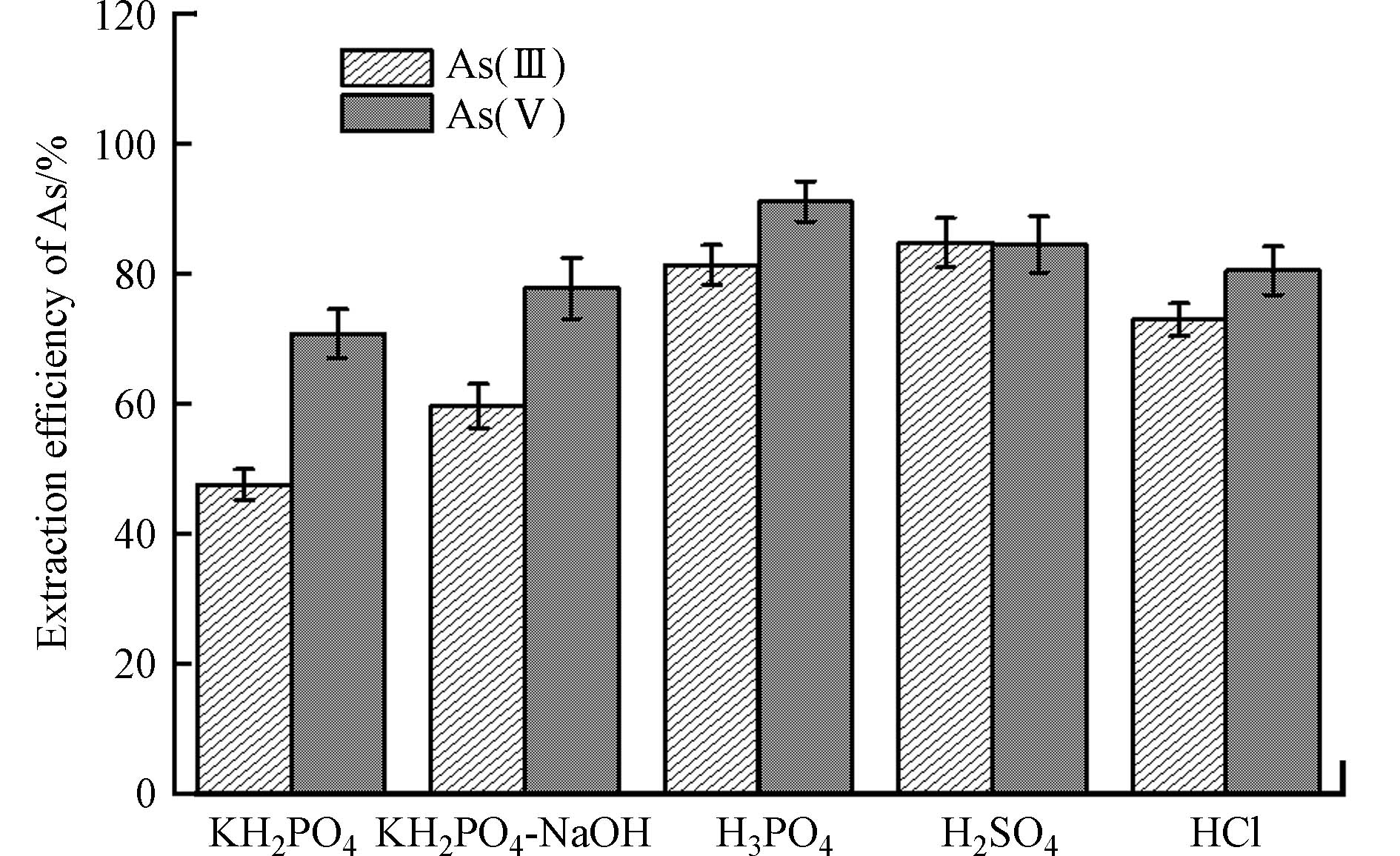
图 2 不同提取剂对NAu-2表面吸附态As(Ⅲ)和As(Ⅴ)的提取效率图
Figure 2. Extraction efficiencies of adsorbed As(Ⅴ) and As(Ⅲ) by different extraction agents
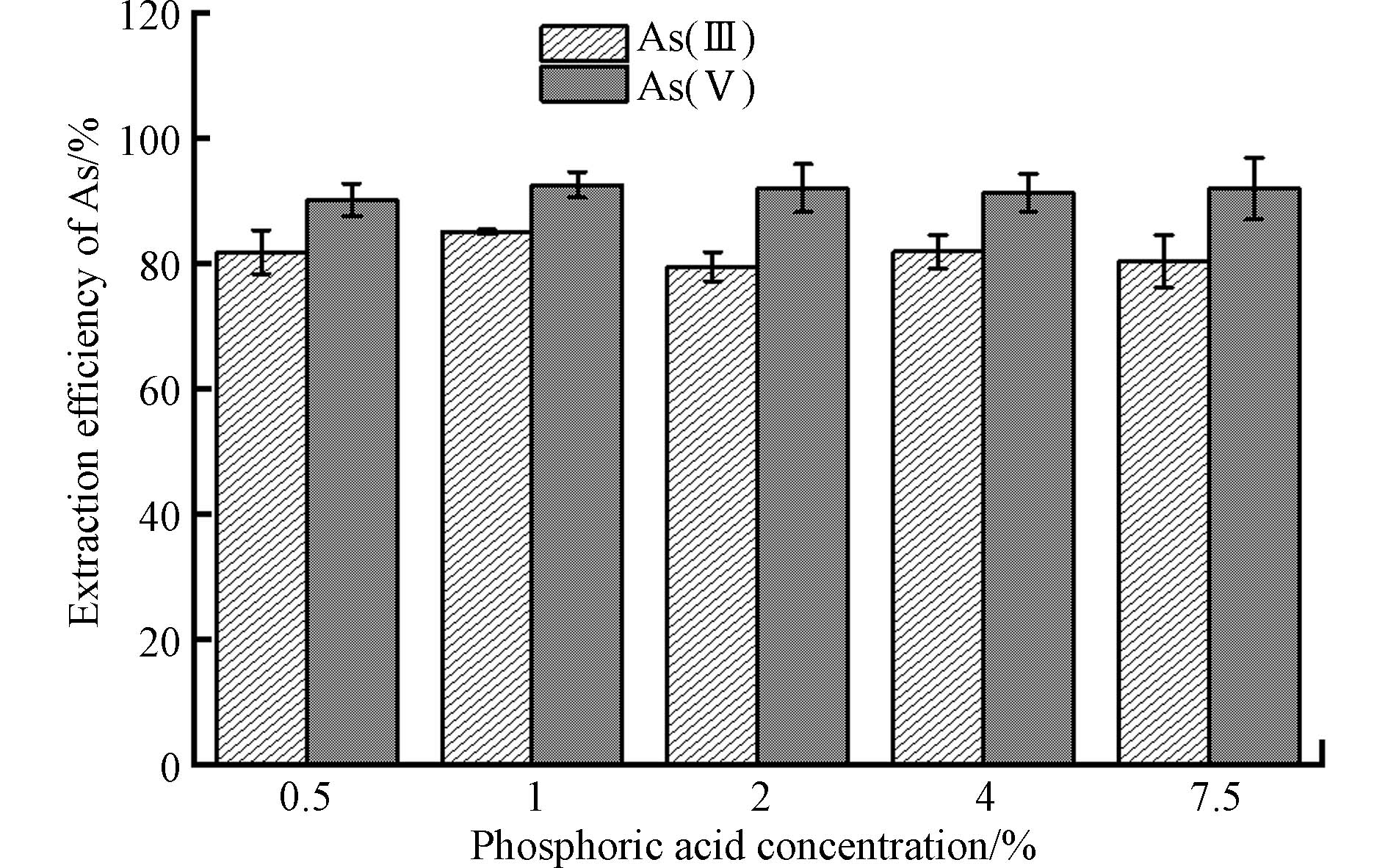
图 3 不同磷酸浓度对As(Ⅲ)和As(Ⅴ)的提取效率
Figure 3. Extraction efficiencies of As(Ⅲ) and As(Ⅴ) under different concentrations of phosphoric acid
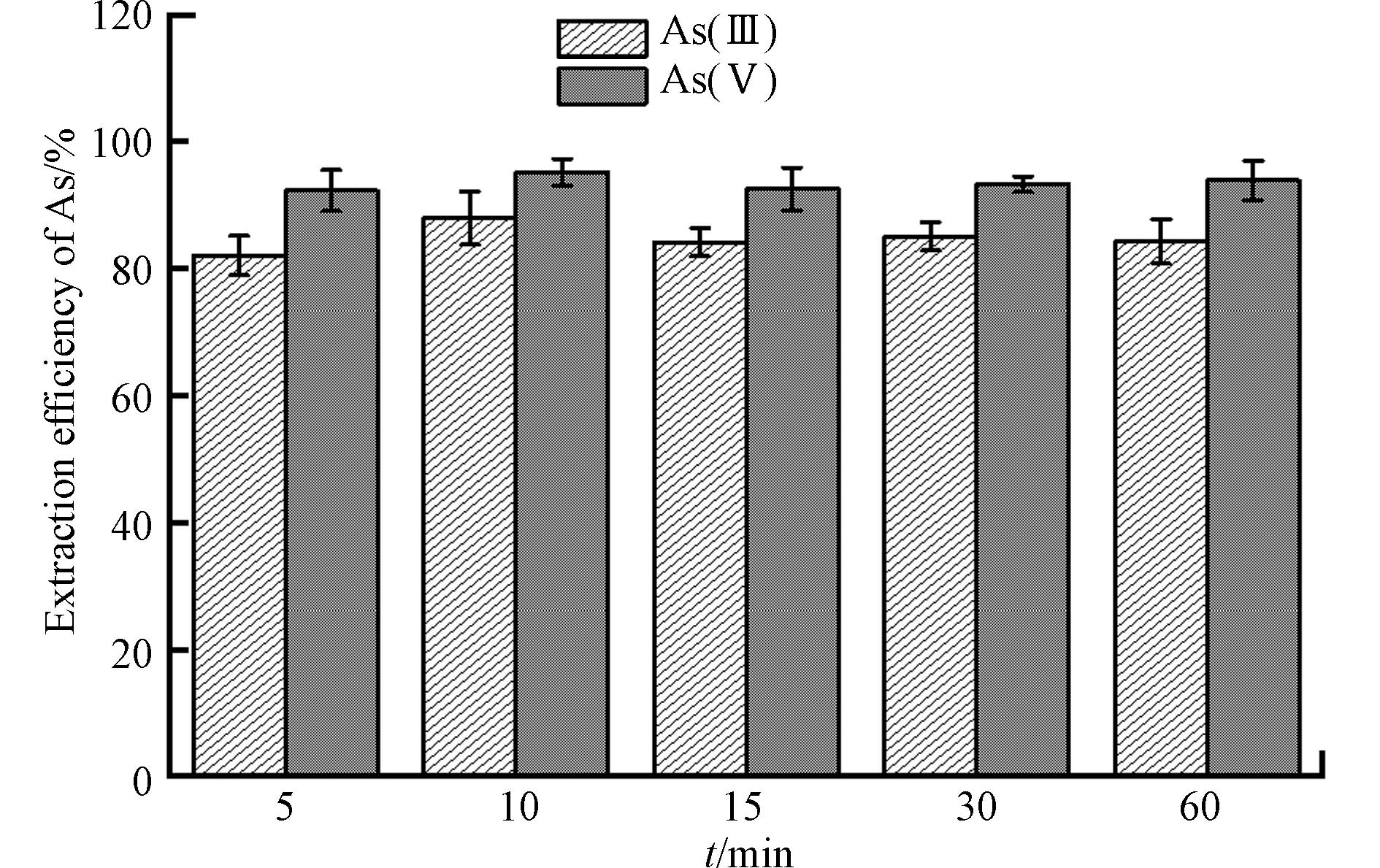
图 4 提取时间对As(Ⅲ)和As(Ⅴ)提取效率的影响
Figure 4. Influence of different extraction time on the extraction efficiencies of As(Ⅲ) and As(Ⅴ)
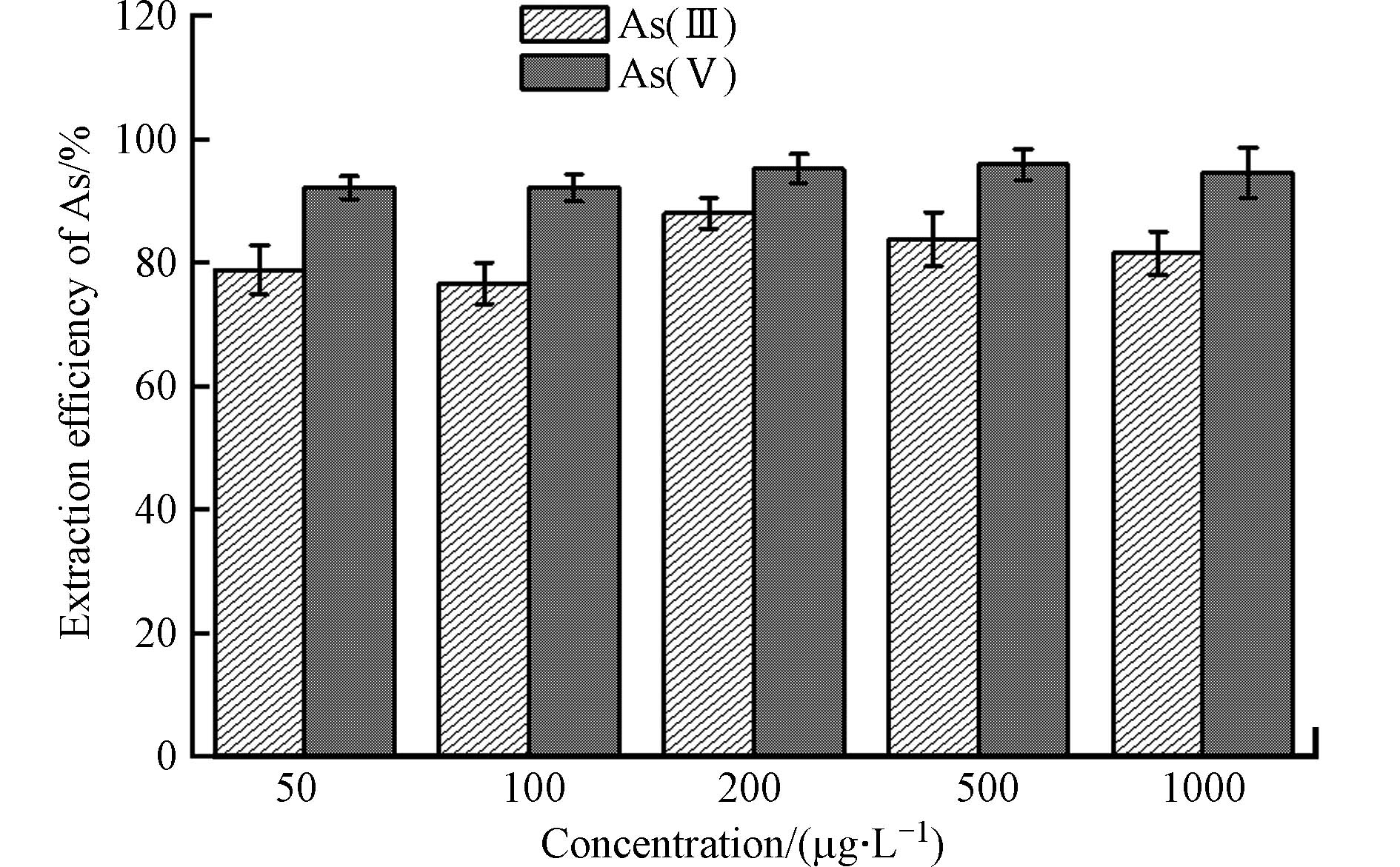
图 5 磷酸对不同浓度As(Ⅲ)和As(Ⅴ)提取效率的影响
Figure 5. Extraction efficiencies of different concentrations of As(Ⅲ) and As(Ⅴ) by phosphoric acid
-
[1] RAHIDUL HASSAN H. A review on different arsenic removal techniques used for decontamination of drinking water[J]. Environmental Pollutants and Bioavailability, 2023, 35(1): 2165964. doi: 10.1080/26395940.2023.2165964 [2] WANG J G, LI Z H, ZHU Q, et al. Review on arsenic environment behaviors in aqueous solution and soil[J]. Chemosphere, 2023, 333: 138869. doi: 10.1016/j.chemosphere.2023.138869 [3] FENDORF S, MICHAEL H A, VAN GEEN A. Spatial and temporal variations of groundwater arsenic in South and Southeast Asia[J]. Science, 2010, 328(5982): 1123-1127. doi: 10.1126/science.1172974 [4] ZAGURY G J, DOBRAN S, ESTRELA S, et al. Inorganic arsenic speciation in soil and groundwater near in-service chromated copper arsenate-treated wood poles[J]. Environmental Toxicology and Chemistry, 2008, 27(4): 799-807. doi: 10.1897/07-305.1 [5] ILGEN A G, KUKKADAPU R K, LEUNG K, et al. “Switching on” iron in clay minerals[J]. Environmental Science: Nano, 2019, 6(6): 1704-1715. doi: 10.1039/C9EN00228F [6] LIN Z, PULS R W. Adsorption, desorption and oxidation of arsenic affected by clay minerals and aging process[J]. Environmental Geology, 2000, 39(7). [7] BOSE P, SHARMA A. Role of iron in controlling speciation and mobilization of arsenic in subsurface environment[J]. Water Research, 2002, 36(19): 4916-4926. doi: 10.1016/S0043-1354(02)00203-8 [8] 罗婷, 景传勇. 地下水砷污染形成机制研究进展[J]. 环境化学, 2011, 30(1): 77-83. doi: 10.1002/etc.362 LUO T, JING C Y. Research progress on mechanisms of arsenic mobilization in groundwater[J]. Environmental Chemistry, 2011, 30(1): 77-83 (in Chinese). doi: 10.1002/etc.362
[9] DONG H. Clay-microbe interactions and implications for environmental mitigation[J]. Elements, 2012, 8(2): 113-118. doi: 10.2113/gselements.8.2.113 [10] GORNY J, BILLON G, LESVEN L, et al. Arsenic behavior in river sediments under redox gradient: A review[J]. Science of the Total Environment, 2015, 505: 423-434. doi: 10.1016/j.scitotenv.2014.10.011 [11] GHORBANZADEH N, JUNG W, HALAJNIA A, et al. Removal of arsenate and arsenite from aqueous solution by adsorption on clay minerals[J]. Geosystem Engineering, 2015, 18(6): 302-311. doi: 10.1080/12269328.2015.1062436 [12] BISHOP M E, GLASSER P, DONG H L, et al. Reduction and immobilization of hexavalent chromium by microbially reduced Fe-bearing clay minerals[J]. Geochimica et Cosmochimica Acta, 2014, 133: 186-203. doi: 10.1016/j.gca.2014.02.040 [13] 张真, 董俊秀, 刘晓雯, 等. 东平湖表层沉积物中砷赋存特征及风险评价[J]. 环境化学, 2020, 39(11): 3190-3199. doi: 10.7524/j.issn.0254-6108.2019082002 ZHANG Z, DONG J X, LIU X W, et al. Arsenic speciation characteristics and risk assessment of surface sediment in Dongping Lake[J]. Environmental Chemistry, 2020, 39(11): 3190-3199 (in Chinese). doi: 10.7524/j.issn.0254-6108.2019082002
[14] LUAN F B, LIU Y, GRIFFIN A M, et al. Iron(III)-bearing clay minerals enhance bioreduction of nitrobenzene by Shewanella putrefaciens CN32[J]. Environmental Science & Technology, 2015, 49(3): 1418-1426. [15] GHORBANZADEH N, LAKZIAN A, HALAJNIA A, et al. Influence of clay minerals on sorption and bioreduction of arsenic under anoxic conditions[J]. Environmental Geochemistry and Health, 2015, 37(6): 997-1005. doi: 10.1007/s10653-015-9708-x [16] CHAPPELL J, CHISWELL B, OLSZOWY H. Speciation of arsenic in a contaminated soil by solvent extraction[J]. Talanta, 1995, 42(3): 323-329. doi: 10.1016/0039-9140(95)01395-R [17] MCLAREN R G, NAIDU R, SMITH J, et al. Fractionation and distribution of arsenic in soils contaminated by cattle dip[J]. Journal of Environmental Quality, 1998, 27(2): 348-354. [18] WENZEL W W, KIRCHBAUMER N, PROHASKA T, et al. Arsenic fractionation in soils using an improved sequential extraction procedure[J]. Analytica Chimica Acta, 2001, 436(2): 309-323. doi: 10.1016/S0003-2670(01)00924-2 [19] WANG J S, TOMLINSON M J, CARUSO J A. Extraction of trace elements in coal fly ash and subsequent speciation by high-performance liquid chromatography with inductively coupled plasma mass spectrometry[J]. Journal of Analytical Atomic Spectrometry, 1995, 10(9): 601-607. doi: 10.1039/ja9951000601 [20] PANTSAR-KALLIO M, MANNINEN P K G. Speciation of mobile arsenic in soil samples as a function of pH[J]. Science of the Total Environment, 1997, 204(2): 193-200. doi: 10.1016/S0048-9697(97)00176-9 [21] THOMAS P, K FINNIE J, G WILLIAMS J. Feasibility of identification and monitoring of arsenic species in soil and sediment samples by coupled high-performance liquid chromatography inductively coupled plasma mass spectrometry[J]. Journal of Analytical Atomic Spectrometry, 1997, 12(12): 1367-1372. doi: 10.1039/a704149g [22] MENG Y, ZHAO Z, BURGOS W D, et al. Iron(III) minerals and anthraquinone-2, 6-disulfonate (AQDS) synergistically enhance bioreduction of hexavalent chromium by Shewanella oneidensis MR-1[J]. Science of the Total Environment, 2018, 640: 591-598. [23] TIAN H, SHI Q, JING C. Arsenic biotransformation in solid waste residue: Comparison of contributions from bacteria with arsenate and iron reducing pathways[J]. Environmental Science & Technology, 2015, 49(4): 2140-2146. [24] LE X C. Arsenic speciation in the environment and humans[M]. Environmental Chemistry of Arsenic, 2001: 115-136. [25] KIM M J, NRIAGU J, HAACK S. Arsenic species and chemistry in groundwater of southeast Michigan[J]. Environmental Pollution, 2002, 120(2): 379-390. doi: 10.1016/S0269-7491(02)00114-8 [26] STAMBERG K, DRTINOVA B, FILIPSKA H, et al. Modelling of acid-base titration curves of mineral assemblages[J]. Open Chemistry, 2016, 14(1): 316-323. doi: 10.1515/chem-2016-0032 [27] 刘冠男, 陈明, 李悟庆, 等. 土壤中砷的形态及其连续提取方法研究进展[J]. 农业环境科学学报, 2018, 37(12): 2629-2638. doi: 10.11654/jaes.2018-0544 LIU G, CHEN M, LI W, et al. A critical review on the speciation and development of sequential extraction procedures for arsenic in soils[J]. Journal of Agro-Environment Science, 2018, 37(12): 2629-2638 (in Chinese). doi: 10.11654/jaes.2018-0544
[28] LARIOS R, FERNÁNDEZ-MATÍNEZ R, RUCANDIO I. Comparison of three sequential extraction procedures for fractionation of arsenic from highly polluted mining sediments[J]. Analytical and Bioanalytical Chemistry, 2012, 402(9): 2909-2921. doi: 10.1007/s00216-012-5730-3 [29] AGHAZADEH V, BARAKAN S, BIDARI E. Determination of surface protonation-deprotonation behavior, surface charge, and total surface site concentration for natural, pillared and porous nano bentonite heterostructure[J]. Journal of Molecular Structure, 2020, 1204: 127570. doi: 10.1016/j.molstruc.2019.127570 [30] JIANG J, XU R K, WANG Y, et al. The mechanism of chromate sorption by three variable charge soils[J]. Chemosphere, 2008, 71(8): 1469-1475. doi: 10.1016/j.chemosphere.2007.12.012 [31] AVENA M J, CABROL R, DE PAULI C P. Study of some physicochemical properties of pillared montmorillonites: Acid-base potentiometric titrations and electrophoretic measurements[J]. Clays and Clay Minerals, 1990, 38(4): 356-362. doi: 10.1346/CCMN.1990.0380404 [32] ZHANG H, SELIM H M. Competitive sorption-desorption kinetics of arsenate and phosphate in soils[J]. Soil Science, 2008, 173(1): 3-12. doi: 10.1097/ss.0b013e31815ce750 [33] MANNING B A, GOLDBERG S. Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals[J]. Soil Science Society of America Journal, 1996, 60(1): 121-131. doi: 10.2136/sssaj1996.03615995006000010020x [34] VIOLANTE A, PIGNA M. Competitive sorption of arsenate and phosphate on different clay minerals and soils[J]. Soil Science Society of America Journal, 2002, 66(6): 1788-1796. doi: 10.2136/sssaj2002.1788 [35] GIRAL M, ZAGURY G J, DESCHÊNES L, et al. Comparison of four extraction procedures to assess arsenate and arsenite species in contaminated soils[J]. Environmental Pollution, 2010, 158(5): 1890-1898. doi: 10.1016/j.envpol.2009.10.041 [36] MARZI M, TOWFIGHI H, SHAHBAZI K, et al. Study of arsenic adsorption in calcareous soils: Competitive effect of phosphate, citrate, oxalate, humic acid and fulvic acid[J]. Journal of Environmental Management, 2022, 318: 115532. doi: 10.1016/j.jenvman.2022.115532 [37] 邹强, 刘芳, 杨剑虹. 紫色土中砷、磷的吸附-解吸和竞争吸附[J]. 应用生态学报, 2009, 20(6): 1383-1389. ZOU Q, LIU F, YANG J H. Adsorption-desorption and competitive adsorption of arsenic and phosphorus in purple soil[J]. Chinese Journal of Applied Ecology, 2009, 20(6): 1383-1389 (in Chinese).
[38] AMSTAETTER K, BORCH T, LARESE-CASANOVA P, et al. Redox transformation of arsenic by Fe(II)-activated goethite (alpha-FeOOH)[J]. Environmental Science & Technology, 2010, 44(1): 102-108. [39] BEDNAR A J, GARBARINO J R, BURKHARDT M R, et al. Field and laboratory arsenic speciation methods and their application to natural-water analysis[J]. Water Research, 2004, 38(2): 355-364. doi: 10.1016/j.watres.2003.09.034 [40] 杨斐, 赵姝婷, 史烨弘, 等. 柠檬酸、EDTA、皂素对砷污染土壤的修复效果研究[J]. 矿冶, 2020, 29(3): 102-105. doi: 10.3969/j.issn.1005-7854.2020.03.020 YANG F, ZHAO S T, SHI Y H, et al. Study on the remediation effect of citric acid, EDTA and saponin on arsenic contaminated soil[J]. Mining and Metallurgy, 2020, 29(3): 102-105 (in Chinese). doi: 10.3969/j.issn.1005-7854.2020.03.020
-
 点击查看大图
点击查看大图
计量
- 文章访问数: 688
- HTML全文浏览数: 688
- PDF下载数: 13
- 施引文献: 0





