

-
挥发性有机物(volatile organic compounds,VOCs)是大气中普遍存在的一类化合物,是形成近地面臭氧和二次有机气溶胶(secondary organic aerosol,SOA)的重要前体物[1]. VOCs可以通过人为源和自然源直接排放,也可以通过大气化学反应二次转化形成[2]. 从全球尺度上看,自然源排放(主要是植被排放)VOCs占主导地位,据估算,其排放通量是人为源的十倍以上[3]. 自然源排放的VOCs以烯烃为主,包括异戊二烯、单萜烯、倍半萜烯和一些氧化物[4]. 与人为源排放的VOCs(主要为芳香族化合物)相比,烯烃结构中含有的不饱和双键使其有更高反应活性,更易发生大气氧化反应并生成复杂的二次有机产物[5]. 由于不饱和碳-碳双键的存在,烯烃可以和O3迅速发生臭氧化反应,生成初级臭氧化产物,而其他烃类如烷烃、多环芳烃、醛类、酮类、醇类一般不与O3反应[6]. 因此,在没有竞争关系的情况下,臭氧分解可能是烯烃最重要去除方式. 鉴于烯烃在大气化学中的重要作用,其臭氧分解反应受到研究人员的广泛关注.
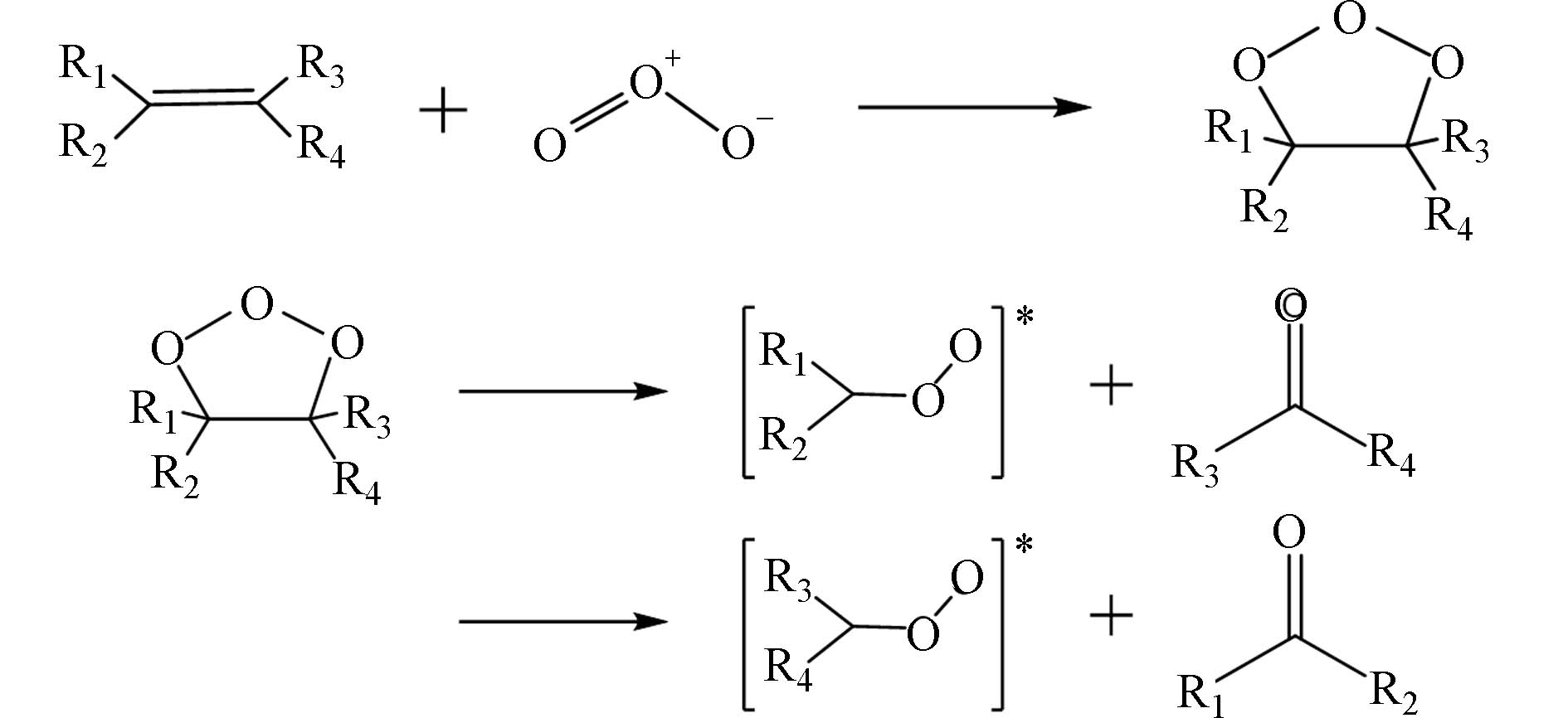
目前,烯烃气相臭氧化反应初始阶段的反应机理已被普遍接受,首先臭氧分子与烯烃的碳碳双键发生1,3-偶极环加成反应生成初级臭氧化物(primary ozonide,POZ),处于激发态的POZ 极不稳定,随即裂解为两对羰基化合物和Criegee中间体(Criegee Intermediate, CI)[7],如图1所示. 该反应是一个自发的放热反应过程,产生的大量热量使CI活化,因此会通过单分子分解、碰撞稳定化和双分子反应等途径释放热量以达到能量上的跃迁[8].
CI是烯烃臭氧化反应最重要的反应产物,其重要性体现在多个方面. 首先CI可以通过分子内的1,4-氢位移生成一个中间产物乙烯基氢过氧化物(vinyl hydroperoxide, VHP),并分解生成羟基自由基(·OH)和乙烯氧自由基(vinyloxyl radical)[9]. 其次CI本身是一个重要的强氧化剂,可以将SO2氧化成H2SO4,而气态H2SO4是二元和三元成核理论的重要组成,可以导致新粒子生成(new particle formation,NPF)事件的发生[10]. 此外,碰撞稳定化的CI(stabilized Criegee Intermediate,SCI)可以和大气中多数气态组分发生双分子反应,生成一系列的二次产物. SCI可以和水反应生成有机酸,后者对于大气酸碱平衡的影响十分重要[11];可以与NO2反应生成硝酸自由基(·NO3),从而影响夜间的大气氧化能力[12];也可以与过氧自由基、氢过氧自由基[13]、有机酸[14]和有机胺[15]反应,生成低聚物,最终导致SOA的生成.
由于中间体短暂的大气寿命(CI约10−9 s,SCI为100—500 ms)[16],在实验室模拟研究中准确测定其动力学参数是十分困难的. 研究需要考虑多种反应过程,例如SCI的直接测定、双分子反应产物识别以及NPF事件发生和颗粒物生长的持续观测,对应的反应时间有毫秒到分钟的跨越. 因此,本实验设计了一个可以通过移动管长来控制反应时间的快速流动反应管.
研究SCI的双分子反应一般要经过两个过程,首先烯烃臭氧化分解生成SCI,其次是SCI与其他物种发生双分子反应. 因为烯烃臭氧化反应有多个中间产物生成,如果两个反应在同一个容器中进行,会互相造成干扰,影响最终的实验结果. 本实验设计了多个鞘层的反应管,有多个反应腔,从而让两个反应独立进行.
此外,SCI活跃的化学性质,可以和大气中绝大多数物种发生双分子反应,因而保证反应体系的纯净是非常必要的. 传统的反应容器“烟雾箱”(chamber)的是由聚四氟乙烯制作而成,虽然材质相对惰性不参与反应过程,但其有较严重的壁效应,可以吸收和释放反应体系中的物种,并且是可逆的[17]. 在反应体系浓度较高的情况下(质量浓度数量级mg·m−3)壁效应是可以忽略的,因为释放的污染物量级小到不会对实验造成干扰[18]. 而SCI的大气平均数浓度较低(约105 cm−3)[19],任何杂质的加入都会影响实验的正常进行. 因此,使用光滑无孔、相对惰性的石英作为反应管的主体材料,既解决了壁效应问题,也可以方便定期清理. 同时,石英良好的光透性对在不同光照条件下的反应研究提供了可能.
快速流动反应管的设计优点之一是在固定的受控条件下(如混合浓度、温度、湿度、压力、光照和反应时间等),在连续流动中产生具有相同物理化学性质的反应产物,另一个优点是反应可以在较短的时间内进行,这样给中间产物的测定和反应过程初期的观测提供了可能. 本文对满足这些标准的流动反应管进行了设计和标定.
-
快速流动反应管总长1.8 m,最大内径为100 mm,主要分为进气模块、反应主体和出气模块三部分,如图所示. 反应主体部分不同直径的4层石英管组合而成,最外层是水套层,提供循环水浴维持反应体系温度恒定. 里三层石英管作为反应容器,由一个固定管和两个可移动伸缩管组成(图2). 通过改变气体流量和管长在毫秒到分钟的范围内调整反应时间. 模块间通过特氟龙密封环密封,两管间通过真空变径接头密封. 在进气模块和出气模块设计了均匀穿孔石英环和六角辐条,该设计保证了气流的定向流动,同时形成了一个湍流区,以便反应物有效混合并快速达到层流状态.
反应管中可以同时进行两个相互独立的反应,两个可移动伸缩管中通入O3和烯烃发生臭氧分解反应,再在固定管中进一步发生SCI双分子反应. 在实验过程中,可通过变更长度、流量、温湿度、光照来改变实验条件,以达到实验目的.
-
快速流动反应管关于流体力学的表征主要通过三个无量纲参数,分别是雷诺数(reynolds number, Re),斯托克斯数(stokes number, Stk)和理查德森数(richardson number, Ri)[20].
-
Re是用来表征流体流动情况的无量纲数,可以衡量气流是否为层流运动或湍流运动[21]. 对于在圆柱型容器流动的气流,Re可以表示为:
其中,ρ为流体密度,ƞ为黏性系数,v为流量,L为特征长度(即流动管内径). 一般来说,23 ℃的气体密度和黏性系数可取经验值分别为1.19 kg·m−3和1.84×10−7 Pa·s. 本实验气体流量变化范围为5—15 L·min−1,算得对应的外管上下限Re分别为51和152,内管上下限Re分别为5和14. 在管流中,一般认为Re小于2000—
2300 的流动是层流,以上计算结果远远小于层流湍流临界值,说明流动管中的反应始终在层流状态进行[22].关于颗粒物的雷诺数定义与气体相似,仅特征长度L变为颗粒物空气动力学直径[23]. 在本实验中,新粒子生成过程中的颗粒物空气动力学直径不超过10 nm,算得的最大Re = 1.6×10−5,说明反应生成的新粒子也是随层流运动.
虽然流体在反应过程中需要保持层流运动,但在反应物接触时需要湍流来充分混合[24]. 流动管中穿孔石英环和六角辐条的设计可以造成湍流混合,并且分散流体快速达到层流效果. 反应物混合需要一定的扩散区域,Bernard利用fluent软件通过网眼计算模拟流体流动路径,发现由湍流点开始的几厘米距离均为扩散混合区域[25]. 本实验气流混合距离约为10 cm,与Bernard结果一致.
-
Stk代表颗粒物停止距离(S)和特征长度(L)的比值,是描述颗粒物在流体流动中行为的无量纲数. Stk > 1,流体绕开障碍物时,颗粒物由于惯性会依然按直线行驶,直至撞上障碍物. Stk < 0.01,流体中的颗粒物会做曲线运动,当流体经过障碍物时,颗粒物随气体同时绕开[21]. 停止距离通过公式(2)计算.
其中,U0为流体流量,ρP为颗粒物密度,dP为颗粒物空气动力学直径,CC为坎宁安滑动校正系数(Cunningham slip correction factor)[26]. 据估算,空气动力学直径为10 nm的颗粒物S = 5×10−8 cm,采样管内径L = 0.65 cm,算得Stk = 7.7×10−8. 这个接近零的值对于非等速取样非常重要. 等速条件要求采样管内的流速U等于自由流速U0,并确保进入采样管的颗粒物浓度C等于稳态颗粒物浓度C0. 当U ≠ U0时,C/C0取决于斯托克斯数,通过经验公式(3)计算[27].
由公式可见,尽管在实验条件下U ≠ U0,但Stk接近0可以确保实验体系里的C/C0 = 1.
-
Ri是表示自然对流和强制对流重要性的无量纲数. 当Ri < 0.1,强制对流起主导作用(流动管内层流),Ri > 10,自然对流占主导地位(温度梯度导致),中间值代表两种对流联合作用[28], 计算见公式(4).
其中,ΔT表示温度差异,g为重力加速度,β空气热膨胀系数(23 ℃时为3.39×10−3 K−1),L为特征长度,U为自由气流流速. 实验通过水浴保持流动管内流体温度恒定,并调节温度范围. 如果管内流体不迅速达到热平衡,那么体系中就会出现温度梯度ΔT,从而引起自然对流. 尽管低Re表示层流运动,由温度变化引起的自然对流还是会导致湍流的发生.
实验通过恒温水浴箱调节温度梯度为10—70 ℃,利用温湿度传感器测量流动管横截面不同位置的温度差异. 以中轴线为圆心,分别测量了最大流速下半径为0、30、45 mm的ΔT,最大ΔT<1 ℃. 通过公式计算Ri<0.1 ,说明流动管内层流状态不随温度变化而变化.
-
在快速流动反应管中,反应物和产物浓度分布受湍流混合的影响. 湍流混合是不同尺度上混合过程的复杂组合,即管半径尺度上的宏观混合、惯性-对流和黏性-对流微观混合以及在分子扩散变得重要的尺度上黏性扩散微观混合[28]. 因此,首先需要标定反应物在不同径向位置的浓度分布.
以臭氧O3为例,通过调节反应管长度为20、30、50、70 cm,测量管截面不同径向采样位置的O3浓度(Model 49i臭氧分析仪, Thermo Scientific)来标定径向混合. 管中总流量为15 L·min−1,管截面的采样位置和臭氧浓度径向分布如图3所示. 可以看出,反应距离为20 cm时管截面不同采样位置的O3浓度波动较大,说明此时并未混合均匀,在30 cm时有一定的波动,> 30 cm时O3达到径向均匀混合. 因此,在后续实验中,设定反应管长 > 30 cm能够获得更加准确的结果. 此外,随着反应管长的增加,O3浓度有微弱的降低,这可能是受流动管壁损失的影响.
-
在动力学实验中,反应物和产物的壁损失对于其浓度平衡有重要影响. 对于相关的动力学计算,忽略反应物的壁损失会导致反应速率系数的高估;而对于产物来说,忽略壁损失则使产物的产率减少. 图3中反应物O3浓度的下降,在单一体系的前提下,可以推测是壁损失所导致的. 由此,通过方程(7)测定了O3在流动管内的一阶反应速率系数.
如图4所示,在流量为15 L·min−1条件下,O3的损失速率为 6.68×10−3 s−1. 由于烯烃臭氧化实验中设定的O3浓度与烯烃相比大大过量(10倍),所以其壁损失可以忽略不计.
产物SCI的扩散限制壁损失的一阶反应系数可以通过公式计算获得[29]:
其中,D是SCI的扩散系数,r代表流动管的内径半径. 假设SCI的扩散系数D为0.01 cm2·s−1,则经过流动管的壁损失速率kwall-loss = 1.46×10−3 s−1. Welz等指出,SCI的大气寿命极短,在反应生成后会快速转化,其热分解速率远远高于壁损失速率[30]. 所以对于动力学分析来说,SCI的壁损失是可以忽略不计.
-
反应速率的计算是动力学研究的一项重要参数,反映了不同物种在大气中的活跃程度及物种间的化学平衡关系. 本实验以2,3-二甲基-2-丁烯(tetramethyl ethene,TME)和O3反应为例,通过以H3O+为试剂离子的高分辨率化学电离飞行时间质谱仪(high resolution time-of-flight chemical ionization mass spectrometer, HToF-CIMS, Aerodyne)[31]实测TME浓度并计算了烯烃臭氧化反应的反应速率系数,并与文献值进行比较.
假设TME和O3反应为二级反应,可以得到以下方程.
由于实验设定O3浓度大大过量,因此其浓度可以当作常数,此时反应速率变成了假一级. 根据式(11),该反应的反应速率系数可以通过改变臭氧浓度和改变反应时间两种方法计算.
改变臭氧浓度的方法是通过固定反应时间,测定不同O3浓度梯度下的TME变化来实现的. 以
$ \left[{\mathrm{O}}_{3}\right] $ 和$ \mathrm{l}\mathrm{n}\dfrac{{\left[\mathrm{T}\mathrm{M}\mathrm{E}\right]}_{t}}{{\left[\mathrm{T}\mathrm{M}\mathrm{E}\right]}_{0}} $ 为坐标轴作线性关系,斜率即为-kt,其反应时间可以通过实验流量15 L·min−1和管径100 mm换算得到. 图5表示了不同管长条件下$ \left[{\mathrm{O}}_{3}\right] $ 和$ \mathrm{l}\mathrm{n}\dfrac{{\left[\mathrm{T}\mathrm{M}\mathrm{E}\right]}_{t}}{{\left[\mathrm{T}\mathrm{M}\mathrm{E}\right]}_{0}} $ 的线性关系,具体结果总结在表1中.反应管长为20 cm和30 cm时,由于反应物未混合均匀,导致计算的反应速率与文献中报道的数值偏差较大,而管长为50—90 cm的计算反应速率与文献值[6]有较好的吻合. 说明在相对较长的时间内流动管反应体系更加稳定,适用于相关反应动力学的研究.
此外,实验还建立了计算反应时间和有效反应时间
$ {t}_{\mathrm{e}\mathrm{f}\mathrm{f}}=1.0274{t}_{\mathrm{c}\mathrm{a}\mathrm{l}}-2.0574 $ 的线性关系(图6). 当反应时间较短时,可以将计算反应时间换算成有效反应时间来进行动力学研究,如果是反应时间相对较长的情况,可以用实验得出的参数直接计算.反应速率系数还可以通过固定臭氧浓度,改变反应时间的方法计算得到. 通过调节实验管长,从而达到改变反应时间的目的. 与改变臭氧浓度的方法类似,以和为坐标轴作线性关系,得到斜率-k[O3],然后代入O3浓度即可求得反应速率系数k.
图7表示了不同臭氧浓度梯度下
$ {t}_{\mathrm{c}\mathrm{a}\mathrm{l}} $ 和$ \mathrm{l}\mathrm{n}\dfrac{{\left[\mathrm{T}\mathrm{M}\mathrm{E}\right]}_{t}}{{\left[\mathrm{T}\mathrm{M}\mathrm{E}\right]}_{0}} $ 的线性关系,计算的反应速率总结在表2中. 可以看出,计算出的反应速率整体比文献值[6]偏大,这可能与反应时间较短,O3浓度分布不均匀有关,臭氧分析仪测得的结果比实际浓度更低. 同时,使用该方法不能够求得有效反应时间$ {t}_{\mathrm{e}\mathrm{f}\mathrm{f}} $ ,以及建立$ {t}_{\mathrm{c}\mathrm{a}\mathrm{l}} $ 和$ {t}_{\mathrm{e}\mathrm{f}\mathrm{f}} $ 的线性关系. 因此,将固定反应时间、改变臭氧浓度的方法应用在动力学计算中更为合适. -
由于烯烃臭氧化反应生成的SCI化学性质非常活跃,可以快速发生单分子热分解,也可以与大气中的污染成分发生双分子反应. 同时,受实验条件的限制,SCI很难被仪器直接观察到. 因此,精确计算不同烯烃臭氧化产生的SCI的产率有很大难度. 本实验通过质子转移反应化学电离质谱仪(proton transfer reaction chemical ionization mass spectrometer, PTR-CIMS)[32]测定烯烃浓度,NO3-为试剂离子的HToF-CIMS[33]测定气态H2SO4浓度,SO2分析仪(Model 43i, Thermo Scientific)测定SO2浓度,从而间接测定SCI产率并与文献值进行比较. 实验以SCI与SO2发生双分子反应生成气态硫酸为理论基础进行.
对于对称烯烃(如TME,臭氧化反应只生成1种SCI)而言,可以假设YSCI+OH=1(Criegee自由基单分子分解会生成·OH自由基,剩余的会碰撞稳定化生成SCI),则YSCI通过方程表示:
其中,
$ {{\mathrm{H}}_{2}{\mathrm{S}\mathrm{O}}_{4}}_{\mathrm{S}\mathrm{C}\mathrm{I}} $ 代表只有SCI与SO2反应生成的硫酸浓度,$ {{\mathrm{H}}_{2}{\mathrm{S}\mathrm{O}}_{4}}_{\mathrm{O}\mathrm{H}+\mathrm{S}\mathrm{C}\mathrm{I}} $ 代表·OH自由基和SCI分别与SO2反应生成的H2SO4总和,根据YSCI+OH=1,比值$ {{\mathrm{H}}_{2}{\mathrm{S}\mathrm{O}}_{4}}_{\mathrm{S}\mathrm{C}\mathrm{I}}/{{\mathrm{H}}_{2}{\mathrm{S}\mathrm{O}}_{4}}_{\mathrm{O}\mathrm{H}+\mathrm{S}\mathrm{C}\mathrm{I}} $ 即为SCI产率.实验前通入N2、O3和SO2,首先先测量流动管中背景H2SO4信号(H2SO4浓度需要将仪器信号通过标定系数转化得到,计算比例时不需要);然后通入·OH自由基清除剂环己烷(去除效率>95%),得到添加清除剂时的背景H2SO4信号;接着添加TME,获得SCI与SO2反应生成的H2SO4信号;最后断开环己烷,能得到·OH自由基和SCI分别与SO2反应生成的H2SO4总和,如图8所示.
根据方程(16),最终算得TME为前体物的SCI产率YTME-SCI =0.353±0.007,这与Berndt[34]和Hakala等[35]的结果都很接近.
在实验过程中,H2SO4的信号值随着SO2浓度的升高不断升高,说明SCI并未完全反应或者SO2和SCI的反应与其他潜在双分子反应存在竞争关系. 当SO2浓度达到很高(约17—20 mg·m-3)时,H2SO4信号值变化减小,趋于平缓,表明SO2与SCI的反应此时已处于主导地位,通过标定后的最大H2SO4浓度,除以反应消耗的烯烃浓度,即为SCI产率. 图9展示了不同烯烃臭氧化体系中H2SO4生成量与SO2浓度的关系,通过计算,得出各体系的SCI产率分别为:雪松烯:61.9%,α-蒎烯:9.5%,β-蒎烯:39.5%,环己烯:6.6%,丙烯:63.5%,1-丁烯:62.2%,与Drozd等[36]的结果相似. 总体来说,链烯烃的SCI产率高于环烯烃,外环烯烃的SCI产率高于内环烯烃,倍半萜的SCI产率高于单萜.
-
烯烃气相臭氧化反应过程会生成化学性质非常活跃的CI,该中间体在大气中会快速发生单分子热分解,也可以与大气中的污染成分发生双分子反应. 由于CI的大气寿命非常短暂,在实验室模拟研究中准确测定其动力学参数是十分困难的. 因此,本实验设计了大气压力快速流动反应管,并对其进行了流体力学表征和动力学标定. 经验证,该流动管具有良好的温度和流量控制功能;可以同时进行两个独立反应(如烯烃+O3和SCI+SO2);可以通过调节长度控制反应时间;易拆卸清洗,最大限度减少可能影响反应的痕量污染物的存在;以及能够提供精确的、直接的动力学信息.
用于烯烃臭氧化反应的快速流动管的设计与动力学研究
Design and kinetic study of fast flow tube for alkene ozonation reaction
-
摘要: Criegee中间体是烯烃气相臭氧化反应过程中最重要的中间产物,可以发生单分子分解生成羟基自由基,也可以与大气中的污染成分发生双分子反应,化学性质十分活跃. 本文设计了大气压力快速流动反应管用于烯烃臭氧化反应及后续二次反应的研究. 该流动管具有良好的温度和流量控制功能;可以同时进行两个独立反应(如烯烃+O3和SCI+SO2);可以通过调节长度控制反应时间;易拆卸清洗,最大限度减少可能影响反应的痕量污染物的存在. 通过3个流体力学无量纲参数雷诺数、斯托克斯数和理查德森数对流动管进行表征,并利用高分辨率化学电离飞行时间质谱对烯烃臭氧化反应及后续双分子反应进行动力学研究. 经验证,新设计的流动管适用于烯烃臭氧化体系的研究,并且能够提供精确的、直接的动力学信息.Abstract: Criegee intermediate is the most important intermediate product in the process of alkene ozonolysis, which can undergo unimolecular decomposition to form hydroxyl radicals or bimolecular reaction with atmospheric pollutants, and has very active chemical properties. In this paper, atmospheric pressure fast flow reaction tube was designed for the study of alkene ozonation reaction and subsequent secondary reactions. The flow tube has good temperature and flow control; two independent reactions (e.g., alkene + O3 and SCI + SO2) can be carried out independently; allows control of the reaction time by adjusting the length; and is easily disassembled for cleaning, minimizing the presence of trace contaminants that may affect the reaction. The flow tube was characterized by three hydrodynamic dimensionless parameters, Reynolds number, Stokes number and Richardson number. Kinetic study of the alkene ozonation reaction and subsequent bimolecular reaction using High Resolution Time-of-Flight Chemical Ionization Mass Spectrometer. The newly designed flow tube was proven to be suitable for the study of alkene ozonation system and to provide accurate and direct kinetic information.
-
Key words:
- flow tube /
- alkene ozonation reaction /
- reaction rate /
- yield.
-
-

图 2 大气压力快速流动反应管示意图
Figure 2. Schematic diagram of atmospheric pressure fast flow reaction tube

图 3 管截面采样位置及O3浓度在不同管长的径向分布
Figure 3. Radial distribution of O3 concentration at different tube lengths
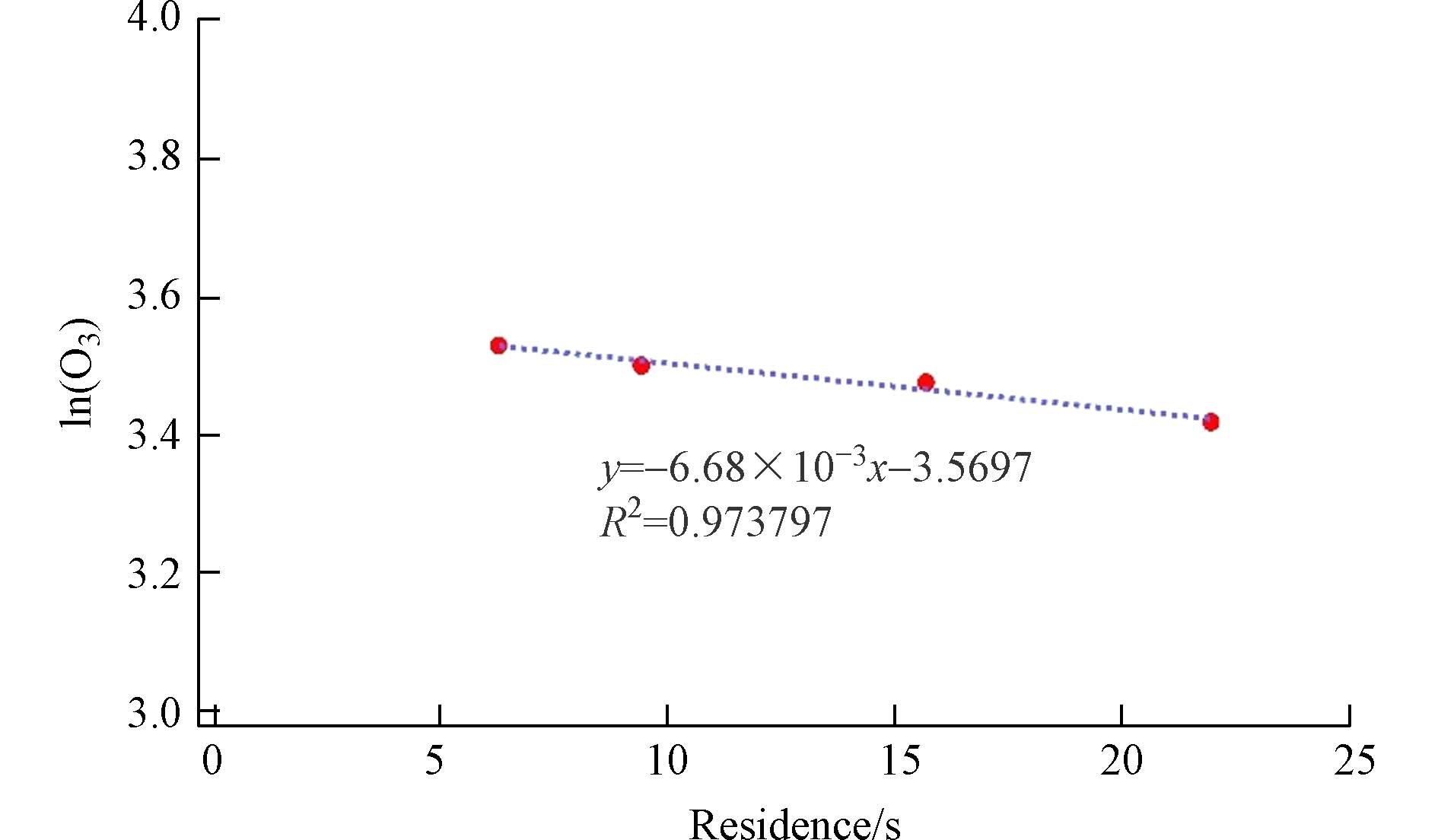
图 4 中心径向采样位置O3的轴向分布
Figure 4. Axial distribution of O3 concentration at the central radial sampling position
图 5 不同管长条件下O3浓度与ln [TME]t/[TME]0的线性关系
Figure 5. O3 concentration vs ln([TME]t/[TME]0) under different tube lengths
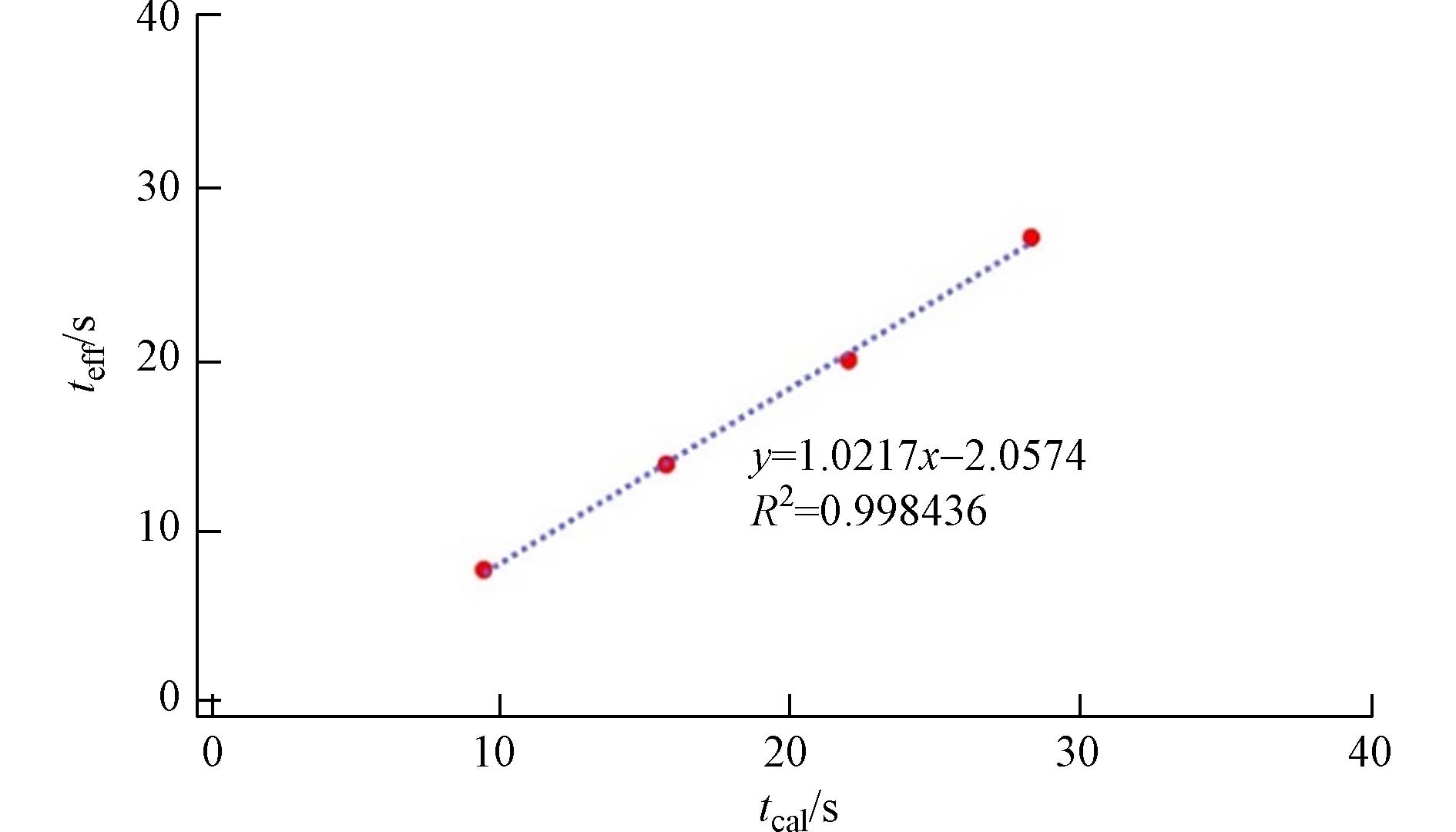
图 6 计算反应时间tcal和有效反应时间teff的线性关系
Figure 6. Correlation of measured reaction time tcal and effective reaction time teff

图 7 不同O3浓度条件下反应时间tcal与ln [TME]t/[TME]0的线性关系
Figure 7. Measured reaction time tcal vs. ln([TME]t/[TME]0) under different tube lengths
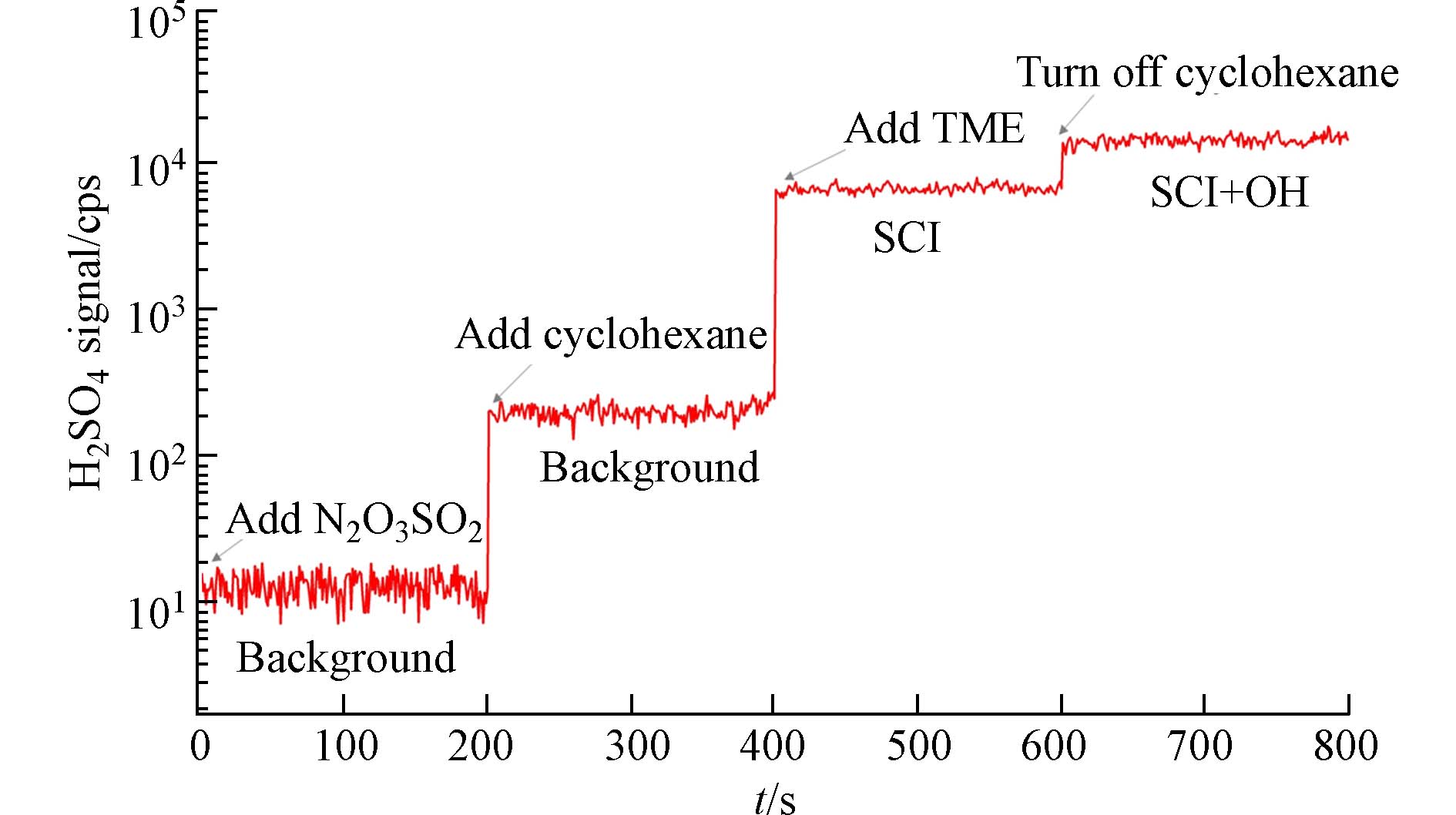
图 8 不同条件下气态H2SO4信号值
Figure 8. Gaseous H2SO4 signal under different reaction conditions
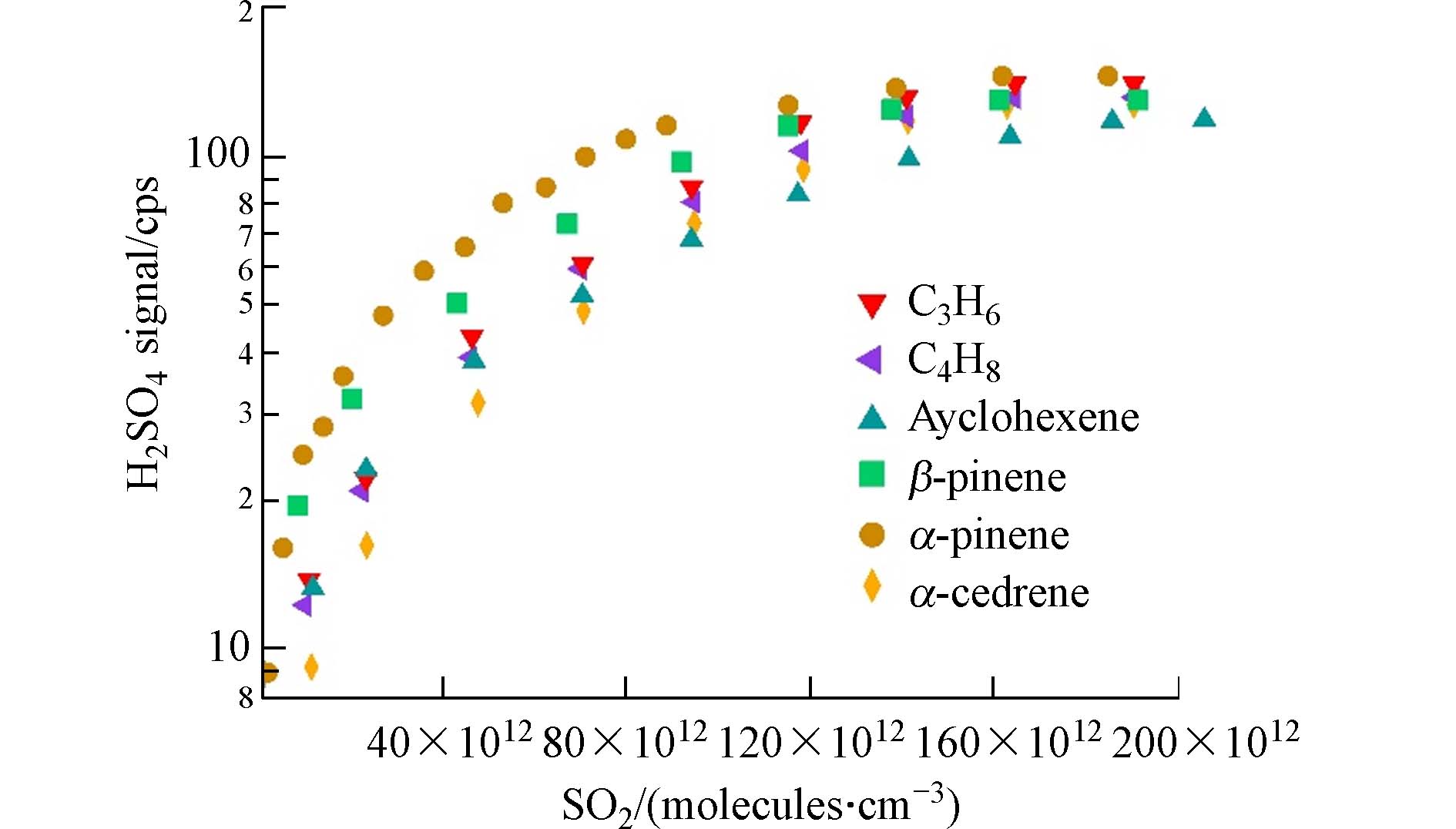
图 9 不同烯烃臭氧化体系中H2SO4生成量与SO2浓度的关系
Figure 9. Relationship between H2SO4 production and SO2 concentration in different alkene ozonation reactions
表 1 不同管长条件下TME与O3反应的动力学参数
Table 1. Kinetic parameters of the reaction between TME and O3 under different tube length
反应管长/cm
Tube length斜率
Slope计算反应时间/s
Calculated reaction
time, tcal计算反应速率/
(cm3·molecule−1·s−1)
Calculated reaction rate, k文献反应速率[6] /
(cm3·molecule−1·s−1)
Literature reaction rate, k有效反应时间/s
Effective reaction
time, teff20 2.51×10−14 6.3 (3.98±0.78)×10−15 (1.13±0.93)×10−15 4.21 30 8.76×10−15 9.4 (0.93±0.14)×10−15 7.75 50 1.57×10−14 15.7 (1.00±0.11)×10−15 13.89 70 2.26×10−14 22.0 (1.03±0.12)×10−15 20.00 90 3.07×10−14 28.3 (1.09±0.11)×10−15 27.17  下载: 导出CSV
下载: 导出CSV
表 2 不同臭氧浓度条件下TME与O3反应的动力学参数
Table 2. Kinetic parameters of the reaction between TME and O3 under different ozone concentration
O3浓度(V/V)
O3 concentrationO3数浓度/(molecule·cm−3)
O3 number concentration斜率
Slope计算反应速率/(cm3·molecule−1·s−1)
Calculated reaction rate, k文献反应速率[6] /(cm3·molecule−1·s−1)
Literature reaction rate, k1.0×10−7 2.46×1012 0.013 (5.28±0.98)×10−15 (1.13±0.93)×10−15 2.0×10−7 4.92×1012 0.016 (3.25±0.41)×10−15 3.0×10−7 7.38×1012 0.021 (2.85±0.35)×10−15 4.0×10−7 9.84×1012 0.022 (2.24±0.29)×10−15 5.0×10−7 1.23×1013 0.032 (2.60±0.30)×10−15
下载: 导出CSV
-
[1] 唐孝炎, 张远航, 邵敏. 大气环境化学-第2版[M]. 北京: 高等教育出版社, 2006. TANG X Y, ZHANG Y H, SHAO M. Atmospheric environmental chemistry-second edition[M]. Beijing: Higher Education Press, 2006(in Chinese).
[2] ATKINSON R. Gas-phase tropospheric chemistry of volatile organic compounds: 1. alkanes and alkenes[J]. Journal of Physical and Chemical Reference Data, 1997, 26(2): 215-290. doi: 10.1063/1.556012 [3] GUENTHER A, HEWITT C N, ERICKSON D, et al. A global model of natural volatile organic compound emissions[J]. Journal of Geophysical Research, 1995, 100(D5): 8873. doi: 10.1029/94JD02950 [4] ATKINSON R, AREY J. Gas-phase tropospheric chemistry of biogenic volatile organic compounds: A review[J]. Atmospheric Environment, 2003, 37: 197-219. doi: 10.1016/S1352-2310(03)00391-1 [5] WENNBERG P O, BATES K H, CROUNSE J D, et al. Gas-phase reactions of isoprene and its major oxidation products[J]. Chemical Reviews, 2018, 118(7): 3337-3390. doi: 10.1021/acs.chemrev.7b00439 [6] ATKINSON R, AREY J. Atmospheric degradation of volatile organic compounds[J]. Chemical Reviews, 2003, 103(12): 4605-4638. doi: 10.1021/cr0206420 [7] CRIEGEE R. Mechanism of ozonolysis[J]. Angewandte Chemie International Edition in English, 1975, 14(11): 745-752. doi: 10.1002/anie.197507451 [8] CALVERT J G. The mechanisms of atmospheric oxidation of the alkenes[M]. New York: Oxford University Press, 2000. [9] FANG Y, LIU F, BARBER V P, et al. Communication: Real time observation of unimolecular decay of Criegee intermediates to OH radical products[J]. The Journal of Chemical Physics, 2016, 144(6): 061102. doi: 10.1063/1.4941768 [10] MAULDIN III R L, BERNDT T, SIPILÄ M, et al. A new atmospherically relevant oxidant of sulphur dioxide[J]. Nature, 2012, 488(7410): 193-196. doi: 10.1038/nature11278 [11] CHAO W, HSIEH J T, CHANG C H, et al. Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor[J]. Science, 2015, 347: 751-754. doi: 10.1126/science.1261549 [12] VEREECKEN L, HARDER H, NOVELLI A. The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere[J]. Physical Chemistry Chemical Physics, 2012, 14(42): 14682-14695. doi: 10.1039/c2cp42300f [13] ZHAO Y, WINGEN L M, PERRAUD V, et al. Role of the reaction of stabilized Criegee intermediates with peroxy radicals in particle formation and growth in air[J]. Physical Chemistry Chemical Physics, 2015, 17(19): 12500-12514. doi: 10.1039/C5CP01171J [14] SAKAMOTO Y, INOMATA S, HIROKAWA J. Oligomerization reaction of the criegee intermediate leads to secondary organic aerosol formation in ethylene ozonolysis[J]. The Journal of Physical Chemistry A, 2013, 117(48): 12912-12921. doi: 10.1021/jp408672m [15] DUPORTÉ G, RIVA M, PARSHINTSEV J, et al. Chemical characterization of gas- and particle-phase products from the ozonolysis of α-pinene in the presence of dimethylamine[J]. Environmental Science & Technology, 2017, 51(10): 5602-5610. [16] OLZMANN M, KRAKA E, CREMER D, et al. Energetics, kinetics, and product distributions of the reactions of ozone with ethene and 2, 3-dimethyl-2-butene[J]. The Journal of Physical Chemistry A, 1997, 101(49): 9421-9429. doi: 10.1021/jp971663e [17] LOZA C L, CHAN A W H, GALLOWAY M M, et al. Characterization of vapor wall loss in laboratory chambers[J]. Environmental Science & Technology, 2010, 44(13): 5074-5078. [18] YAO L, MA Y, WANG L, et al. Role of stabilized Criegee Intermediate in secondary organic aerosol formation from the ozonolysis of α-cedrene[J]. Atmospheric Environment, 2014, 94: 448-457. doi: 10.1016/j.atmosenv.2014.05.063 [19] NOVELLI A, HENS K, TATUM ERNEST C, et al. Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument[J]. Atmospheric Chemistry and Physics, 2017, 17(12): 7807-7826. doi: 10.5194/acp-17-7807-2017 [20] EZELL M J, CHEN H H, ARQUERO K D, et al. Aerosol fast flow reactor for laboratory studies of new particle formation[J]. Journal of Aerosol Science, 2014, 78: 30-40. doi: 10.1016/j.jaerosci.2014.08.009 [21] HINDS W C. Aerosol technology: properties, behavior, and measurement of airborne particles[M]. 2nd ed. New York: Wiley, 1999 [22] PAVELYEV A A, RESHMIN A I, TEPLOVODSKII S K, et al. On the lower critical Reynolds number for flow in a circular pipe[J]. Fluid Dynamics, 2003, 38(4): 545-551. doi: 10.1023/A:1026369727130 [23] HUMPHRY K J, KULKARNI P M, WEITZ D A, et al. Axial and lateral particle ordering in finite Reynolds number channel flows[J]. Physics of Fluids, 2010, 22(8): 081703. doi: 10.1063/1.3478311 [24] KHALIZOV A F, EARLE M E, JOHNSON W J W, et al. Modeling of flow dynamics in laminar aerosol flow tubes[J]. Journal of Aerosol Science, 2006, 37(10): 1174-1187. doi: 10.1016/j.jaerosci.2005.11.008 [25] BERNARD F, FEDIOUN I, PEYROUX F, et al. Thresholds of secondary organic aerosol formation by ozonolysis of monoterpenes measured in a laminar flow aerosol reactor[J]. Journal of Aerosol Science, 2012, 43(1): 14-30. doi: 10.1016/j.jaerosci.2011.08.005 [26] SEINFELD J H, PANDIS S N. Atmospheric chemistry and physics: from air pollution to climate change[M]. 2nd ed. Hoboken, NJ: Wiley, 2006. [27] BELYAEV S P, LEVIN L M. Techniques for collection of representative aerosol samples[J]. Journal of Aerosol Science, 1974, 5(4): 325-338. doi: 10.1016/0021-8502(74)90130-X [28] KHALIZOV A F, EARLE M E, JOHNSON W J W, et al. Development and characterization of a laminar aerosol flow tube[J]. Review of Scientific Instruments, 2006, 77(3): 033102. doi: 10.1063/1.2175958 [29] SIPILÄ M, JOKINEN T, BERNDT T, et al. Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids[J]. Atmospheric Chemistry and Physics, 2014, 14(22): 12143-12153. doi: 10.5194/acp-14-12143-2014 [30] WELZ O, SAVEE J D, OSBORN D L, et al. Direct kinetic measurements of criegee intermediate (CH2 OO) formed by reaction of CH2I with O2[J]. Science, 2012, 335(6065): 204-207. doi: 10.1126/science.1213229 [31] ZHENG J, MA Y, CHEN M D, et al. Measurement of atmospheric amines and ammonia using the high resolution time-of-flight chemical ionization mass spectrometry[J]. Atmospheric Environment, 2015, 102: 249-259. doi: 10.1016/j.atmosenv.2014.12.002 [32] MA Y, DIAO Y W, ZHANG B J, et al. Detection of formaldehyde emissions from an industrial zone in the Yangtze River Delta region of China using a proton transfer reaction ion-drift chemical ionization mass spectrometer[J]. Atmospheric Measurement Techniques, 2016, 9(12): 6101-6116. doi: 10.5194/amt-9-6101-2016 [33] ZHENG J, YANG D S, MA Y, et al. Development of a new corona discharge based ion source for high resolution time-of-flight chemical ionization mass spectrometer to measure gaseous H2SO4 and aerosol sulfate[J]. Atmospheric Environment, 2015, 119: 167-173. doi: 10.1016/j.atmosenv.2015.08.028 [34] BERNDT T, JOKINEN T, SIPILÄ M, et al. H2SO4 formation from the gas-phase reaction of stabilized Criegee Intermediates with SO2: Influence of water vapour content and temperature[J]. Atmospheric Environment, 2014, 89: 603-612. doi: 10.1016/j.atmosenv.2014.02.062 [35] HAKALA J P, DONAHUE N M. Pressure-dependent criegee intermediate stabilization from alkene ozonolysis[J]. The Journal of Physical Chemistry A, 2016, 120(14): 2173-2178. doi: 10.1021/acs.jpca.6b01538 [36] DROZD G T, DONAHUE N M. Pressure dependence of stabilized criegee intermediate formation from a sequence of alkenes[J]. The Journal of Physical Chemistry A, 2011, 115(17): 4381-4387. doi: 10.1021/jp2001089 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1228
- HTML全文浏览数: 1228
- PDF下载数: 25
- 施引文献: 0













