

-
生活垃圾焚烧、再生有色金属冶炼、铁矿石烧结等典型工业热过程是无意生成持久性有机污染物(unintentionally produced persistent organic pollutants,UPOPs)的重要排放源[1 − 2],如多氯代二苯并-对-二噁英(PCDDs)、多氯代二苯并呋喃(PCDFs)、多氯联苯(PCBs)、多氯萘(PCNs)等. 尽管工艺技术和烟气污染控制设施的完善使得烟气中二噁英的脱除效率达98%以上[3],但由于二噁英的源头生成量大,其排放浓度仍难以达到大气污染物的排放标准[4],由此推测,其他UPOPs的生成量和排放量必然相当可观,因此,从源头控制UPOPs的生成才是其减排的关键. 了解UPOPs的生成机制对于其源头控制至关重要. 研究表明,工业热过程中其他UPOPs与二噁英具有相似的生成机制[5]. 二噁英在燃烧反应中主要通过高温气相合成和低温异相催化合成[6]. 高温气相合成发生的温度范围为500—800 °C,氯源和碳源在气相中合成二噁英,通常气相中生成二噁英需要相关前驱体参与,如氯苯、氯酚等,或者通过其他脂肪烃缩合而成[7 − 8]. 低温异相催化合成发生的温度区间为200—400 °C,包括了前驱体(precursors)生成和从头合成(de novo). 前驱体生成途径是指氯苯、氯酚、多环芳烃等前驱体被吸附在颗粒物表面,然后在过渡金属催化剂的参与下生成二噁英,如图1所示. 从头合成是指碳源在氯源、金属催化剂的参与下生成二噁英. 然而,现有UPOPs生成机制的研究主要是基于稳定中间产物和目标UPOPs的检测,对于自由基中间体的研究非常少.
在热反应中,有机物的生成转化通常是自由基参与的反应[11]. 自由基具有高度活性,存留时间短至10−9 s,常见的瞬时自由基包括氢自由基(·H)、羟基自由基(·OH)、超氧阴离子自由基(·O2―)等[12]. 近年来,有研究者在大气颗粒物中检测到了环境持久性自由基(environmentally persistent free radicals,EPFRs)[13]. 不同于活性强、寿命短的瞬时自由基,环境持久性自由基具有更高的稳定性和持久性,其寿命可达几小时到几个月,甚至几年[14],这是因为有机自由基与金属氧化物之间发生了电子转移,形成了更加稳定的化学结构,能够在环境中长时间存在. 目前,常见的EPFRs主要有苯氧类自由基、半醌类自由基和环戊二烯类自由基等,这些EPFRs普遍稳定存在于土壤、沉积物和大气细颗粒物(PM)等环境介质中[15]. EPFRs通过环境介质进入人体组织后,能够诱发产生活性氧自由基,导致人体细胞被氧化,产生细胞毒素,造成DNA损伤和细胞死亡[16]. 土壤中存在的EPFRs还会抑制农作物的发芽和生长[17]. 因此,EPFRs被认为是一类新环境有害污染物[18]. 除了在环境介质中存在,在工业燃烧产生的飞灰中也能检测到EPFRs[19]. EPFRs容易在燃烧系统和其他热过程的后燃烧区以及烟气冷却区大量生成[20],而这部分也是UPOPs生成的主要区域,由于EPFRs与UPOPs具有相似的生成条件和区域,因此,有研究提出,EPFRs是UPOPs生成的重要自由基中间体[21].
前期研究提出前驱体、金属催化剂、反应气氛、温度等是影响二噁英等UPOPs生成的关键因素. 以二噁英为例,以氯酚或氯苯为前驱体更容易生成PCDDs,而以多环芳烃或焦炭作为前驱体则更容易生成PCDFs[22]. 250—450 °C是二噁英生成的最佳温度,在该温度范围内,随着氧气浓度从0%增加到21%,二噁英的生成量也随之增加,且PCDDs的增量大于PCDFs[23]. 金属氯化物比金属氧化物的催化活性更强,不同金属化合物对氯代多环芳烃生成的促进能力排序为CuCl2 > FeCl3 > FeCl2 > CuO > Fe2O3 > PbCl2 > Blank > ZnCl2 > PbO > ZnO[24]. 而添加一些钙基、氮基和硫基阻滞剂能够抑制二噁英的生成[25 − 26]. 作为UPOPs生成潜在的自由基中间体,目前尚未有研究总结这些因素对EPFRs生成的影响. 本文总结了热反应过程中前驱体、反应气氛、温度、金属催化剂对EPFRs生成的影响,论述了生成二噁英等UPOPs过程中可能存在的持久性自由基转化机制,并结合现有UPOPs的源头阻滞技术对今后UPOPs与EPFRs协同抑制进行了展望.
-
目前,EPFRs主要通过电子顺磁共振技术(electron paramagnetic resonance,EPR)进行检测和甄别. 理论上,可以通过EPR谱图的g值来鉴别EPFRs是属于碳中心还是氧中心自由基. 通常,自由基中的未成对电子越靠近氧原子,其g值越大. 前期研究总结了不同类型自由基的g值,其中自由电子的g值等于
2.0023 ;碳中心自由基的g值小于2.003(石墨碳自由基g值等于2.0028 [27];多环芳烃自由基g值约为2.0026 );以碳为中心、邻位有氧原子的自由基g值范围为2.003—2.004[20];氧中心自由基g值大于2.004[28]. g值也与自由基上取代基团(氯取代基和羟基)的数量有关,氯取代的自由基在低温下具有较高的g值,且随着温度升高发生脱氯反应,g值也随着降低. 与此同时,EPFRs的结构还会由于与反应基质的相互作用发生改变,从而进一步改变其EPR谱图.然而,实际工业飞灰中存在的有机物和颗粒物组分十分复杂,一方面,飞灰中存在的磁性物质会影响电子顺磁共振波谱仪的稳定性;另一方面,难以通过单一的g值去推测多组分有机自由基的结构. 因此,实际工业样品中EPFRs的定性和定量分析是比较困难的. 为了更方便地鉴定热过程中生成的EPFRs类型,并分析它们与UPOPs之间的关联性,许多研究者采用单一的金属氧化物与惰性基质混合,利用单一的前驱体,如氯酚、氯苯、多环芳烃等典型有机物,在基质上模拟工业热过程中EPFRs和UPOPs的生成反应. 在热反应过程中,EPFRs的生成和转化受多种因素的影响,不同工况下、不同前驱体在不同金属氧化物上生成EPFRs的情况见表1.
-
不同的前驱体由于化学结构和取代基种类、位置和数量的不同,会经历不同的反应路径,从而生成不同种类的EPFRs,如图2所示. Dellinger课题组[29 − 30]对比了对苯二酚、邻苯二酚、苯酚、2-氯酚、一氯苯、1,2-二氯苯在金属氧化物表面生成的自由基种类,尽管不同的前驱体都能生成苯氧自由基或半醌自由基,但是它们的反应路径并不一致. 苯酚上只有一个羟基取代基,与金属氧化物表面反应时通过H2O的脱除生成苯氧自由基,通过密度泛函理论(DFT)计算可知这个苯氧自由基上的未成对电子可以在氧原子上或碳原子上,即氧中心或者碳中心的自由基. 对苯二酚与苯酚一样,通过H2O的脱除生成对半醌自由基,根据g值为
2.0062 ,说明生成的是氧中心自由基. 1,2-二氯苯和邻苯二酚在相邻的碳上有两个取代基,在金属氧化物表面上,氯取代基或羟基通过HCl或H2O的脱除,生成苯氧自由基或半醌自由基,两个邻位的取代基也可能同时化学吸附在金属氧化物表面,生成邻半醌自由基. 1,2-二氯苯可以脱除一个或两个HCl生成2-氯苯氧自由基或邻半醌自由基,由于受邻位上氯原子的影响,2-氯苯氧自由基的g值比无氯取代的苯氧自由基的g值大. 同样,邻苯二酚可以通过一个或两个H2O的脱除生成苯氧自由基或邻半醌自由基,但是从能量的角度来看,同时脱除两个H2O更容易,因此,会优先生成邻半醌自由基,继而通过表面介导反应进一步降解成苯氧自由基. 一氯苯与1,2-二氯苯、邻苯二酚一样,在低温下生成半醌自由基,而在高温下同时生成半醌自由基和苯氧自由基,这是因为一氯苯在CuO上反应时会首先生成1,2-二氯苯,然后再按照二氯苯的反应路径生成自由基. 不同的自由基具有不同的活性,通过DFT计算,邻半醌自由基比对半醌自由基更容易生成,且更不容易被分解或氧化,苯氧自由基比半醌自由基更难生成,但是更容易被分解或氧化[20]. 然而,目前大多数研究所使用的前驱体都是有取代基团的芳香烃,D’Arienzo等[33]采用无取代基的苯作为前驱体,利用CuxO作为反应基质,通过计算模型并结合实验验证,发现O2分子在一价铜的激活下形成了过氧化物,苯在这个过氧化物的氧化下形成了苯氧自由基,因此,对于无取代基的前驱体,氧的嵌入反应是后续生成EPFRs的重要步骤. -
不同的反应气氛会影响生成自由基的浓度和种类. 在惰性气氛中,由于氮气等惰性气体不会与自由基反应,因此,自由基主要是通过有机物的分解而生成. 而氧气很容易参与自由基反应,在反应中会生成过氧化物自由基和其他新自由基,导致g值增加[51],因此氧气在自由基的反应中具有重要作用. 在不同氧气浓度下,1,2,3-三氯苯在CuO和Fe2O3表面主要生成两种类型自由基,随着氧气浓度从0%增加至20%,碳中心自由基的浓度减少,而氧中心自由基浓度增加[50]. 2-氯酚在氧化条件下生成的氯醌浓度比在惰性气氛下的多,而醌类化合物的生成通常伴随有半醌类自由基的产生,因此,可以推测2-氯酚在氧化条件下生成的半醌类自由基浓度也更高[45]. 在氧化性能力更高的臭氧气氛下,多环芳烃生成的自由基浓度更高,且淬灭得快,这说明在臭氧气氛下,自由基的反应活性更强,生成的自由基类型主要有三类,其中两类属于半醌类自由基,还有一类属于碳中心的多环芳烃类自由基[34]. 不同类型的自由基在不同气氛下的反应活性也有差异,在惰性气氛下,半醌自由基和苯氧自由基不容易被氧化,环戊二烯自由基也不容易热分解,而在氧气气氛下,半醌自由基和苯氧自由基比环戊二烯自由基更稳定,而环戊二烯自由基更容易被氧化[20].
-
温度会对EPFRs的浓度和种类产生影响. 在CuO上,对苯二酚和邻苯二酚在50—100 °C时能生成稳定的自由基,但在这个温度区间自由基的浓度没有明显增加,随着温度升高至300 °C,自由基的浓度明显增加,且g值逐渐稳定在2.006—2.007之间. 而氯苯在低于150 °C时没有出现自由基信号,直到225 °C时自由基的浓度才明显增加,在相对较低温度时,自由基的g值较高,为2.007—2.008,而随着温度升高,g值降至
2.0045 —2.0055 [20]. 1,2,3-三氯苯在金属氧化物表面反应时,随着温度升高,生成的自由基浓度逐渐增加,在350 °C时达到最高. 在250—450 °C时生成的自由基类型主要为氯代苯氧自由基,而当温度升高至550 °C时,生成了氧中心的氯代邻半醌自由基[50]. 2,4-二氯-1-萘酚在CuO表面反应时,在77 °C时就出现了自由基信号,生成的自由基为氯代萘氧自由基,随着温度升高至300 °C,发生了脱氯反应,生成了萘氧自由基[35]. 苯酚在室温下就容易在纳米ZnO上生成苯氧自由基,且与250 °C下生成的自由基类型一致[40]. 相比之下,羟基取代的前驱体更容易在室温或低温下生成自由基,而只有氯取代基的前驱体则需要在一定的高温条件下才能生成自由基. 不同种类的前驱体在不同温度下生成EPFRs的路径也不一致. 2-氯酚在低温下通常只生成2-氯-苯氧自由基,因为从能量的角度低温下脱除H2O分子比脱除HCl分子更容易,但是在高温下会同时生成2-氯-苯氧自由基和邻半醌自由基. 相反,对于邻苯二酚,低温下在金属氧化物表面同时脱除两个H2O分子从能量的角度看是更容易的,因此只生成一个邻半醌自由基,而在高温下邻半醌自由基则会分解生成苯氧自由基,同样的反应路径也适用于1,2-二氯苯和一氯苯[29]. 不同的自由基在不同温度下也表现出不同的稳定性,在热解条件下,半醌自由基和苯氧自由基在700 °C以下能稳定存在,超过这个温度则会分解,而环戊二烯自由基在相同温度下稳定性更强[20]. -
目前,EPFRs仅在颗粒物表面被检测到,因此表面介导的反应可能是EPFRs的主要生成途径. 通常,有机前驱体首先化学吸附在金属氧化物表面,发生电子转移,生成EPFRs并激发后续的反应. 金属氧化物的种类、浓度和颗粒大小会显著影响EPFRs的生成和稳定性. 对苯二酚、邻苯二酚、苯酚、2-氯酚、1,2-二氯苯在CuO[29]和Fe2O3[30]颗粒表面都生成了苯氧自由基和半醌自由基,有机前驱体化学吸附在金属氧化物表面并向金属氧化物转移了电子,导致金属阳离子被还原,而有机物则生成了自由基. 这些前驱体在CuO、Fe2O3、NiO[31]和ZnO[32]上生成的EPFRs稳定性不同,在ZnO表面生成的EPFRs更稳定,如半醌自由基,寿命可达3—73 d. 苯酚、2-氯酚、1,2-二氯苯在不同浓度的CuO颗粒表面都生成了g值为
2.0037 —2.006的苯氧自由基和半醌自由基. CuO浓度为1%—3%时,苯酚和2-氯酚生成的EPFRs浓度最高大,而1,2-二氯苯生成的EPFRs浓度则随着铜含量的降低而增加,与此同时,自由基的类型也从半醌自由基转变为氯苯氧自由基[44]. 相比于在CuO表面,苯酚、一氯苯和1,2-二氯苯在PbO表面生成的EPFRs寿命更长,可达1—4 d. 苯酚通过羟基中O—H键的均裂生成苯氧自由基,而氯苯和1,2-二氯苯则通过Cl原子的脱除分别生成苯基自由基和氯苯自由基,通过DFT计算,氯苯自由基与PbO颗粒之间的结合能更强,因此它的寿命在这3种自由基中是最长的[43]. 不同粒径的金属氧化物颗粒也会影响EPFRs的生成,纳米级别的CuO、Al2O3、ZnO、NiO颗粒比相应的微米颗粒更能促进EPFRs的生成,这是因为纳米颗粒具有更大的比表面积,能吸附更多的有机自由基并防止了自由基之间的耦合[35].同样的前驱体在不同的金属氧化物上生成的EPFRs种类和浓度有所区别. 金属离子的氧化性越强,越能促进EPFRs的生成,因此,CuO、Al2O3、ZnO、NiO促进2,4-二氯-1-萘酚生成EPFRs的能力顺序为Al2O3 > ZnO > CuO > NiO[35]. 苯酚在ZnO表面主要生成氧中心的苯氧自由基,而在TiO2、CuO和Fe2O3上生成的是碳中心和氧中心苯氧自由基的结合,相比之下,TiO2最能促进苯酚生成EPFRs,这是因为TiO2颗粒中生成了很多超氧化物,能促进有机前驱体的氧化,而苯酚在Fe2O3上生成的EPFRs浓度最低,这是因为Fe2O3的催化活性强,分解了生成的EPFRs[38]. 有机物和不同金属氧化物反应时电子转移的方向是不一致的,对于CuO、Fe2O3、Al2O3、TiO2等大多数金属氧化物,电子转移方向均为有机物向金属离子,有研究通过光电发射技术,进一步证明了苯酚向Fe2O3颗粒的电子转移[36]. 然而,对于ZnO,尽管苯酚在其表面也生成了苯氧自由基,但是通过DFT计算发现电子转移的方向是从ZnO到苯酚[40],反应路径如图3推测. 因此,简单的氧化还原反应并不能统一解释有机前驱体与所有金属氧化物之间的反应,电子传递的方向与金属氧化物的性质相关.
-
目前,低温异相催化合成反应是研究UPOPs生成的自由基机制的主要手段,通过对反应后稳定化合物的检测和反应过程中EPFRs的表征,并结合量子化学计算,能够从分子层面推测从典型前驱体到UPOPs的生成路径,有助于完善UPOPs的生成机制. 现有的研究大多选用有取代基团的芳烃作为前驱体,如氯酚、氯苯等,在CuO、Fe2O3等典型过渡金属催化剂上进行实验研究. 2-氯酚在CuO颗粒表面会生成氧中心的一氯代苯氧自由基,两个一氯代苯氧自由基通过类乌尔曼反应脱水耦联生成4,6-二氯二苯并呋喃(4,6-DCDF). 与此同时,自由基氧原子上的未成对电子会转移到邻位碳上,形成碳中心的苯氧自由基,并与超氧根阴离子结合,在铜表面生成C6H3ClOOH,这个物质会进一步与2-氯酚分子反应,通过脱除两个HCl分子生成无氯取代的二苯并-对-二噁英(DD)[45, 52]. 2-氯酚在Fe2O3上除了生成4,6-DCDF和DD,高温下还生成了4-氯代二苯并呋喃(4-MCDF)和二苯并呋喃(DF),这可能是2-氯酚在高温下首先通过H2O和HCl的脱除生成了邻半醌自由基,邻半醌自由基通过表面缩合反应生成苯氧自由基,然后通过两个苯氧自由基耦合反应生成DF或与一个2-氯酚分子耦合生成4-MCDF[48]. 1,2-二氯苯与2-氯酚的反应路径类似,在CuO表面上首先通过一个HCl分子的脱除生成一氯代苯氧自由基,然后通过自由基-自由基缩合生成4,6-DCDF,后续生成DD的反应路径则与2-氯酚的一致[47]. 2,3,6-三氯酚在CuO上可能生成3种类型的自由基,但通过EPR检测,生成的自由基类型主要为三氯苯氧自由基,即通过H2O的脱除生成. 因此,三氯苯氧自由基后续通过二聚、邻位氯脱除、Smiles重排、闭环、环内氯脱除等五个基本反应生成PCDDs[49]. 其他多氯代苯和多氯代酚生成PCDD/Fs的自由基反应路径可参照图4. DFT计算表明,苯氧自由基比半醌自由基在热力学上更易发生自二聚化反应,因此苯氧自由基形成二噁英的活性要高于半醌自由基[53]. 除了生成PCDD/Fs,氯酚产生的氯代苯氧自由基二聚体还能脱除2个羰基生成二氯-9,10-二氢富瓦烯中间体,继而生成PCNs[5].
氯取代反应是生成二噁英的关键自由基反应步骤. 1,2,3-三氯苯在金属氧化物表面首先通过一个或两个HCl的脱除分别生成二氯苯氧自由基或一氯邻半醌自由基,两个自由基在界面上通过自由基-自由基缩合反应生成二氯代二苯并呋喃(DCDF)、三氯代二苯并呋喃(TrCDF)和四氯代二苯并呋喃(TeCDF),然后再进一步氯化生成高氯代的PCDFs[50]. 而在2-氯酚生成二噁英的模拟实验中,EPR检测到的有机自由基信号峰-峰宽(ΔHp-p)为4,通过量子化学计算预测了可能由2-氯酚产生的65种有机自由基的超精细分裂常数,发现三至四氯代的半醌自由基和2、4、6位氯取代的酚氧自由基与实际EPR谱图匹配度最高,由此推测,在该反应中,氯首先在有机自由基上发生取代反应,继而缩合生成高氯代的二噁英,如图5所示,符合产物中五至八氯代的二苯并-对-二噁英分布较多的规律[54]. 从以上研究可知,在高氯代二噁英生成的自由基反应中,氯是首先在有机自由基上发生取代反应生成高氯代的自由基中间体过渡态,还是在生成的低氯代二噁英上发生逐级取代反应,仍有争议.
母体多环芳烃也是生成UPOPs的重要前驱体,不同于氯代芳烃类前驱体,母体多环芳烃上没有取代基团,也没有氧原子,因此在生成二噁英的过程中氯取代反应和外部氧嵌入反应是重要步骤. 研究发现蒽的分解产物通过重组可以生成二噁英[55]. 进一步分析发现蒽在CuCl2催化下的产物包含了一氯蒽、蒽酮、蒽醌、氯代蒽醌等,结合EPR谱图,推测在反应中生成的持久性自由基类型为蒽类、蒽醌类自由基,氯代蒽醌进一步分解、重组生成二噁英等UPOPs,并且发现反应过程中EPFRs的浓度与UPOPs的浓度呈正相关[56]. 然而,相比于以氯代芳烃为前驱体生成UPOPs的研究,目前以母体多环芳烃为典型前驱体的研究较少. 母体多环芳烃是有机物不完全燃烧的主要产物,掌握多环芳烃生成UPOPs的自由基机制对于完善UPOPs的生成机制具有重要意义. 此外,目前仅有的UPOPs生成的自由基机制的研究主要集中在PCDD/Fs上,缺少对PCBs、PCNs、溴代二噁英等其他UPOPs生成机制的研究,这对烟气污染物减排技术实施过程中多种UPOPs协同脱除的实现具有理论指导意义.
-
在工业热过程中除了通过袋式除尘器、活性炭吸附等污染物控制装置对燃烧后烟气中的二噁英进行末端脱除,还会通过反应工况的优化或者在燃烧前预混区以及燃烧后尾部区喷加阻滞剂的方式[57],以破坏二噁英的生成条件,从而达到源头抑制二噁英生成的目的. 常用的阻滞剂主要有碱性阻滞剂、硫基阻滞剂、氮基阻滞剂等[58],不同阻滞剂均对UPOPs有较好的抑制效果,但是它们的作用机制有所区别,如表2所示.
碱性阻滞剂主要是通过吸收氯源、降低金属氯化物的催化活性来达到抑制UPOPs生成的效果[60, 67],目前研究较多的是CaO,CaO能将CuCl2转化为CuO,并吸收Cl2和HCl,通过吸收氯源和降低催化活性抑制氯苯的生成. CaO的抑制效果受较多因素影响,当Ca/Cl≥17.7时,250 °C下CaO对焚烧飞灰中的PCDD/Fs脱除效率达到87.2%[68]. 在800 °C时,CaO也可以有效抑制17种2,3,7,8-PCDD/Fs的生成[69],且CaO对于六氯代PCDD/Fs的抑制效果最为明显,但不会明显改变PCDD/Fs同类物的分布. 而有研究发现CaO对于PCDFs的降解效果明显好于PCDDs,虽然总PCDD/Fs浓度降低了,但是PCDDs的浓度却略有升高,特别是低氯代PCDDs[70]. 对于不同前驱体(五氯酚和六氯苯)生成二噁英的体系,CaO对PCDDs和PCDFs的抑制效果不同,但是在280—450°C下抑制率均超过了90%[71]. 氯含量也是影响阻滞效果的因素,额外无机氯源(NaCl和CuCl2)的添加也会明显降低钙基阻滞剂的抑制效果[72]. 除了对氯源的吸收和金属催化剂的钝化,CaO对前驱体(五氯酚)还有一定的吸附和分解作用,能通过酸碱中和反应(Acid-base reactions)生成苯氧化钙((PhO)2Ca),从而阻滞UPOPs生成[26]. 也有研究发现尽管二噁英被分解了,但是平均氯化度并未降低,因此CaO对二噁英的抑制并非是一个连续的脱氯反应,而是通过C—O键的断裂降解成了小分子的氯酚、氯苯类结构,因此分解作用占主导地位[73, 74]. 此外,DFT计算表明,CaO能促进苯环的羟基和氯取代,从而抑制了氯苯和氯酚的缩聚[75].
硫基阻滞剂主要是通过释放SO2,与过渡金属催化剂反应生成硫酸盐,从而使催化剂中毒[76],在生活垃圾焚烧过程中添加硫对四氯代至七氯代二苯并呋喃的抑制率高达70%[69]. 氮基阻滞剂则主要通过释放NH3,与Cl2或HCl反应生成NH4Cl,从而削弱有机氯的生成[64]. 在铁矿石烧结过程中添加0.05%的尿素对二噁英的抑制率可达63%以上,但是添加量超过0.5%时反而无抑制作用[77]. 氮硫复合阻滞剂则结合了两者特点,例如硫脲,在240—400 °C之间会逐渐分解为SO2和NH3,使金属催化剂中毒的同时削弱氯在芳烃上的取代反应[78]. 有研究比较了硫脲、4-甲基硫代氨基脲、硫酸铵等多种硫、氮基阻滞剂在铁矿石烧结中对PCBs的抑制效率,发现硫脲的抑制效率最高,达91.4%,这与S+N与Cl的摩尔比相关,(S+N)/Cl摩尔比越大,抑制效果越好[79]. 然而,使用硫氮基类阻滞剂可能会增加烟气后续脱硫脱硝的净化压力[80, 81],若掌控不好添加量和温度反而会促进UPOPs的生成[61, 82].
根据上述研究可知,不同阻滞剂对UPOPs的抑制机制存在区别,主导抑制作用不明,且在抑制过程中很少涉及对自由基中间体的研究. 有研究在蒽生成UPOPs的反应中添加了CaO,发现CaO对EPFRs的抑制率可达85%,对PCDD/Fs、PCBs、PCNs的抑制率分别为97%、93%、97%,实现了EPFRs和UPOPs的协同抑制[56],但并未阐明CaO对持久性自由基的抑制机制. 目前仅有研究发现在模拟飞灰中添加钙和硫能通过抑制过渡金属表面活性位点,限制EPFRs的生成[83],也有研究推测钙基复合氧化物能与酚类前驱体产生的苯氧基结合,从而抑制生成PCDD/Fs的乌尔曼反应[84]. 由此可见,对EPFRs起抑制作用的化合物与二噁英的阻滞剂具有相似的元素组成和作用机制. 由此推测,常用的二噁英阻滞剂也能同时抑制EPFRs的生成. EPFRs作为UPOPs生成反应的重要自由基中间体,其生成转化行为对UPOPs的生成或抑制具有重要作用,然而,现有的研究成果并未充分考虑阻滞剂在抑制UPOPs生成过程中对自由基中间体的抑制作用.
-
掌握工业热过程中UPOPs的生成机制是实现其减排的基础. 目前对UPOPs生成机制的研究主要基于反应前后稳定同类物的检测,缺乏对自由基中间体的认知. 本文对工业热过程中UPOPs生成的自由基机制进行了综述,结合影响UPOPs生成的关键因素,首先讨论了前驱体、反应气氛、温度和金属催化剂对EPFRs的影响. 其次,根据稳定中间产物的结构结合DFT计算,推测了EPFRs的结构以及EPFRs转化生成UPOPs的反应路径,有助于从分子层面完善UPOPs的生成机制. 由于EPFRs也被认为是一类新环境污染物,本文在最后还依据现有UPOPs的阻滞剂和可能的EPFRs抑制机理,展望了EPFRs与UPOPs协同抑制技术. 主要的结论如下:
(1)热过程中EPFRs的生成受温度、气氛、前驱体和金属催化剂的影响,这些因素也是影响UPOPs生成的关键因素. 苯环上不同官能团的分解温度不同,随着温度升高,氯酚类前驱体中的O—H键先断裂,生成苯氧自由基,继而C—Cl键断裂,生成半醌自由基,而无取代基团的前驱体则需要外部氧的嵌入才能生成苯氧自由基. 碳中心自由基在氧气气氛下容易被氧化生成氧中心自由基,因此在氧化性气氛下,氧中心自由基比碳中心自由基更加稳定. 过渡金属的氧化性越强,前驱体与金属之间越容易发生电子转移,促进EPFRs的生成.
(2)目前的研究主要通过前驱体异相催化合成反应结合量子化学计算来推测UPOPs生成的自由基机制. 氯酚在金属氧化物上脱除一个H2O生成氯代苯氧自由基或者脱除H2O和HCl生成半醌自由基,自由基两两聚合生成PCDD或者PCDF,DFT计算表明苯氧自由基比半醌自由基更容易发生自二聚化反应. 而以多环芳烃为前驱体生成UPOPs的自由基机制研究较少.
(3)钙基、硫基和氮基阻滞剂通过削弱有机氯的生成和降低金属催化剂的活性来抑制二噁英等UPOPs的生成,而飞灰中钙和硫的存在能够限制EPFRs的生成,因此,利用二噁英阻滞剂在理论上能协同抑制EPFRs的生成,但这一点还未得到研究论证.
现有UPOPs生成的自由基机制的研究仍存在许多限制和不足. 有机物的热转化反应较复杂,反应中不止存在一种有机自由基,目前很难对有机自由基的EPR谱图进行解卷积分析. 此外,受有机自由基本身超精细耦合常数较小的影响,目前大部分含苯环有机自由基的EPR信号均为一个大的对称峰,少有超精细分裂峰,因此,无法利用EPR谱图对有机自由基的结构直接定性. 有机自由基的准确甄别能为现有UPOPs的生成转化机制提供直接证据. 通过对稳定分子中间产物的表征结合DFT计算能够初步推测生成的有机自由基结构,但该方法难以得到实验验证. 考虑到同位素标记和非同位素标记的有机结构具有不同的EPR超精细谱图,因此,通过对前驱体进行同位素标记来示踪其转化反应,并对有机自由基进行原位检测,能够甄别反应过程中主要的有机自由基中间体.
工业热过程中无意生成持久性有机污染物的自由基转化机制
Free-radical mediated formation mechanisms of unintentionally produced persistent organic pollutants in industrial thermal processes
-
摘要: 掌握无意生成持久性有机污染物(unintentionally produced persistent organic pollutants,UPOPs)的生成机制对于其在典型工业热过程中的源头控制具有重要意义,现有对二噁英等UPOPs生成机制的研究主要是基于反应后稳定化合物和目标同类物分布的检测,缺乏对自由基中间体的研究. 环境持久性自由基(environmentally persistent free radicals,EPFRs)是一类新的环境污染物,其半衰期可达数月,可在环境样品以及二噁英生成过程中检出. 近年来EPFRs被认为是UPOPs生成的重要自由基中间体. 本文对UPOPs生成过程中前驱体种类、金属化合物、温度、反应气氛等对EPFRs生成特性的影响作用进行了综述,分析了前驱体生成UPOPs过程中同种或不同种EPFRs的转化路径. 此外,基于现有UPOPs源头阻滞技术,提出了UPOPs与EPFRs协同控制的潜在方法.Abstract: It is of great significance to master the formation mechanisms of unintentionally produced persistent organic pollutants (UPOPs) for its source control in typical industrial thermal processes. Existing studies on the formation mechanism of UPOPs, e.g. dioxins, are mainly based on the detection of stable compounds and target congeners after reaction, but the detection of free-radical intermediates is lacking. Environmentally persistent free radicals (EPFRs) are a new class of environmental pollutants with a half-life up to several months. They can be detected in environmental samples and dioxin formation processes. EPFRs have been recognized as important free-radical intermediates for UPOPs generation in recent years. In this paper, the critical effects of precursor species, metal compounds, temperature and reaction atmosphere on the formation characteristics of EPFRs are reviewed. The conversion pathways of various EPFRs to UPOPs from different precursors are analyzed. In addition, based on the current UPOPs control technology, a potential method for synergistic control of UPOPs and EPFRs is proposed.
-
-
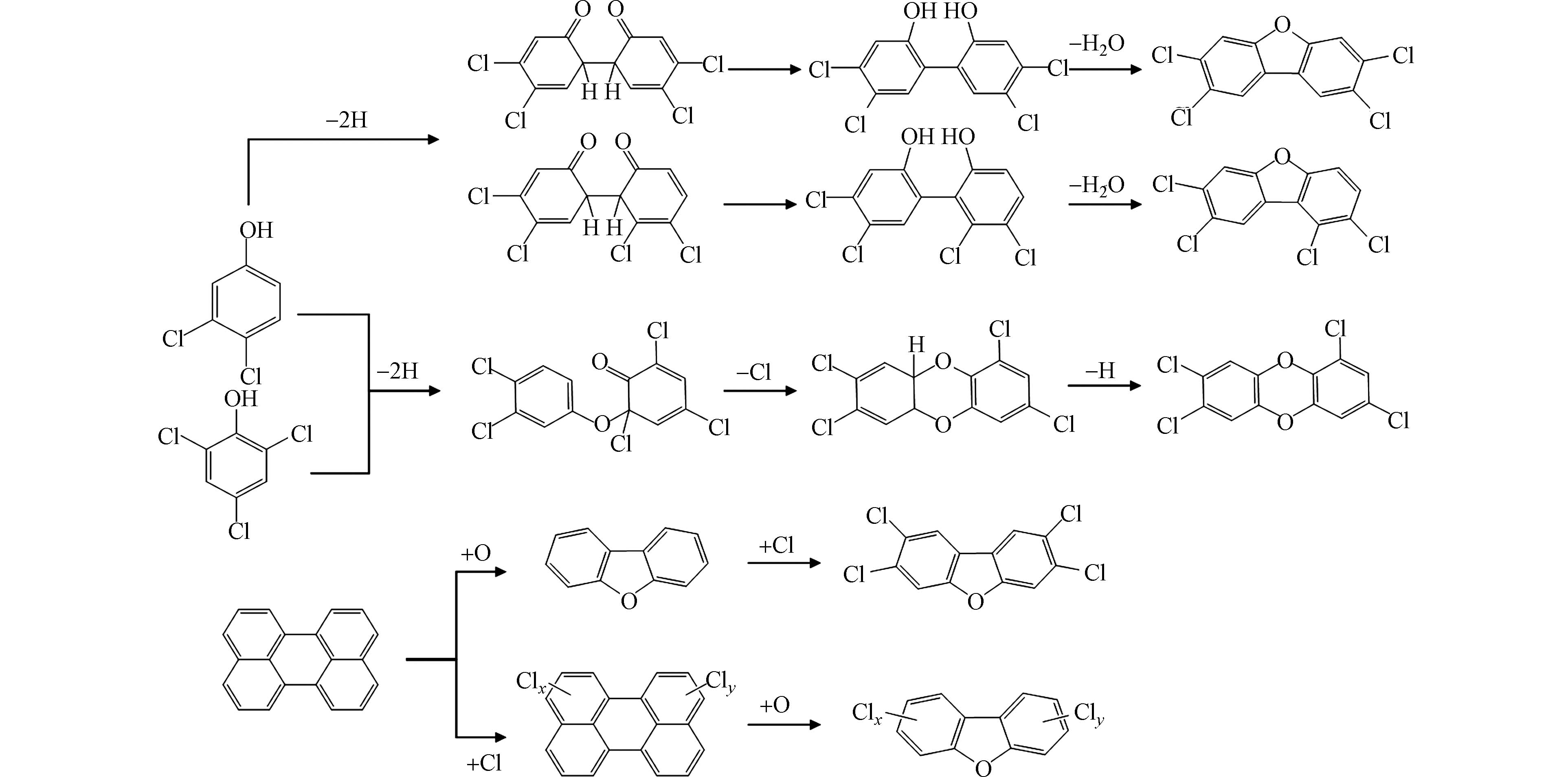
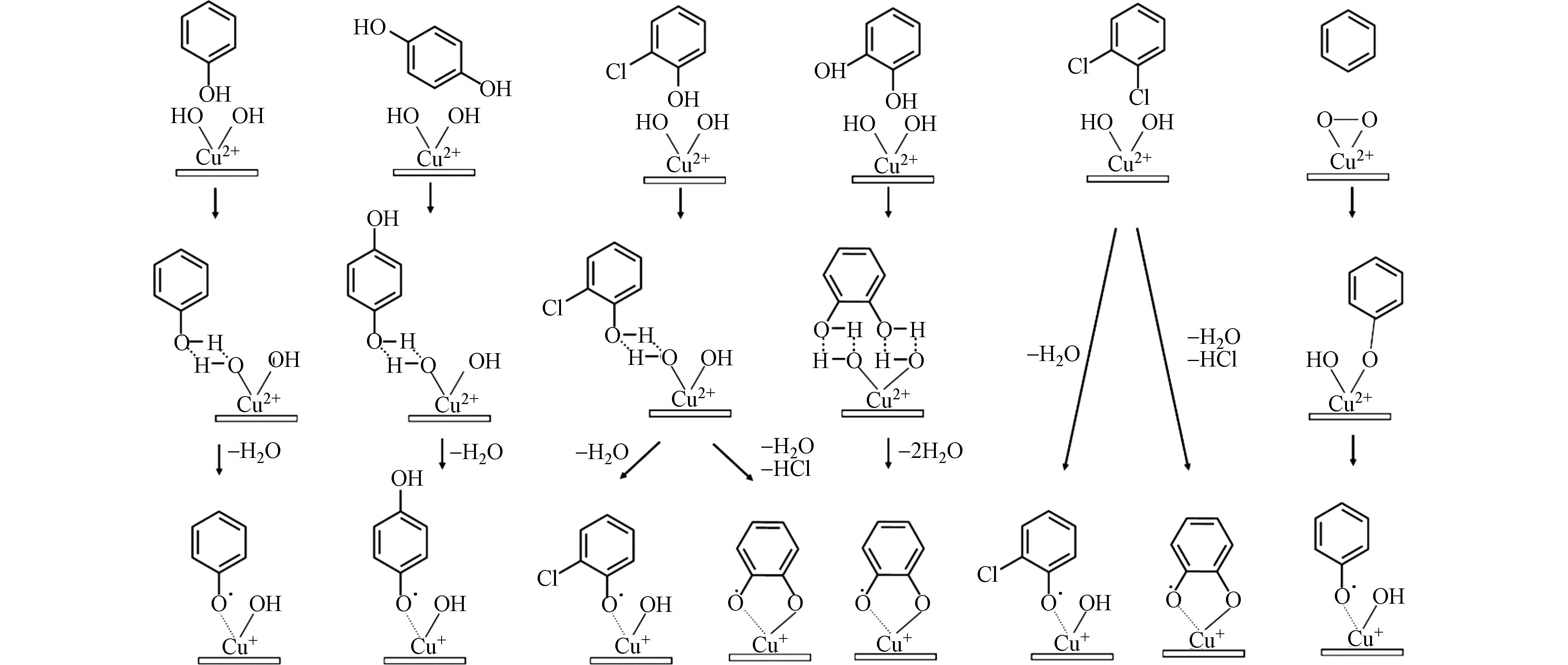
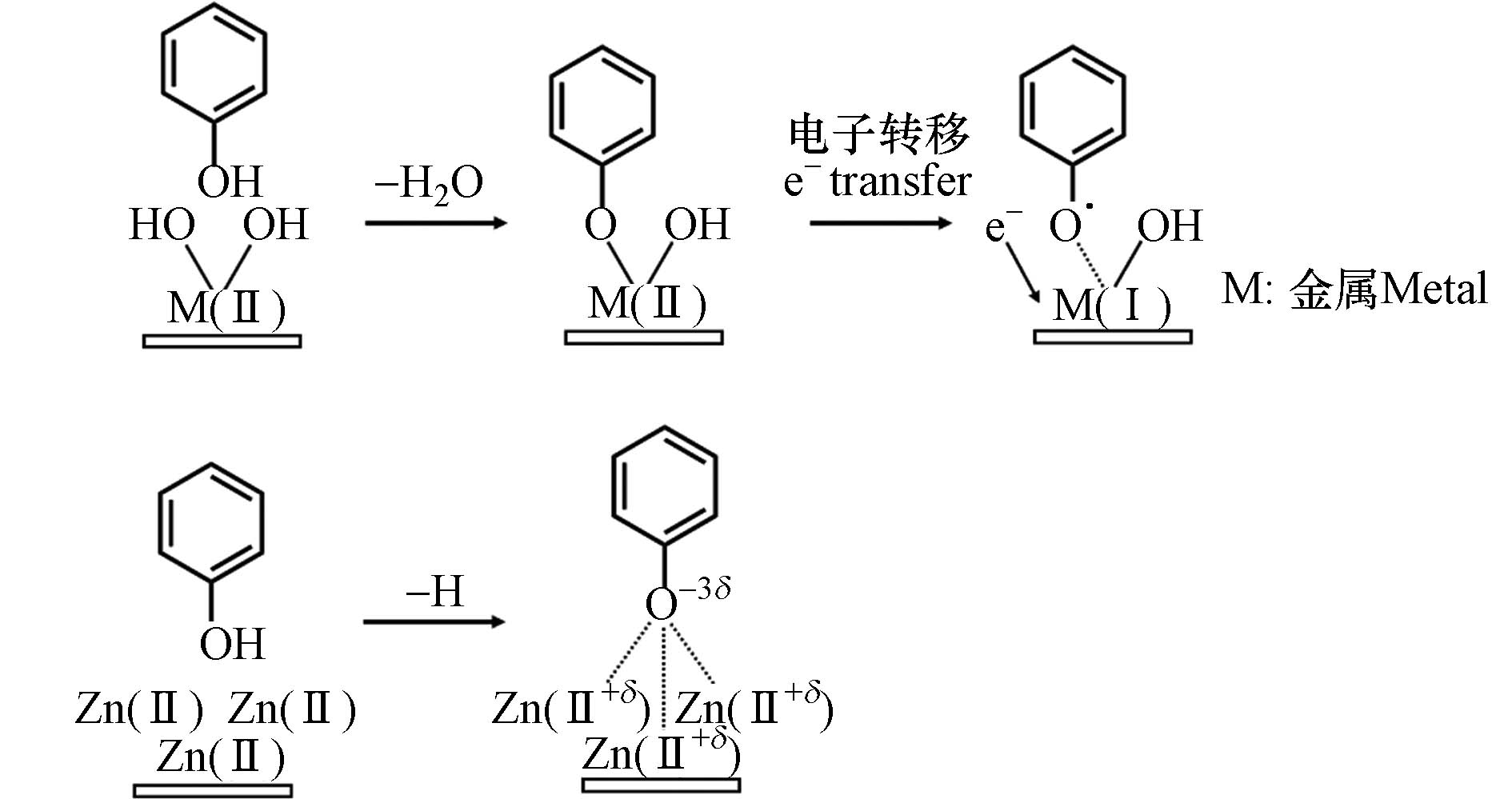
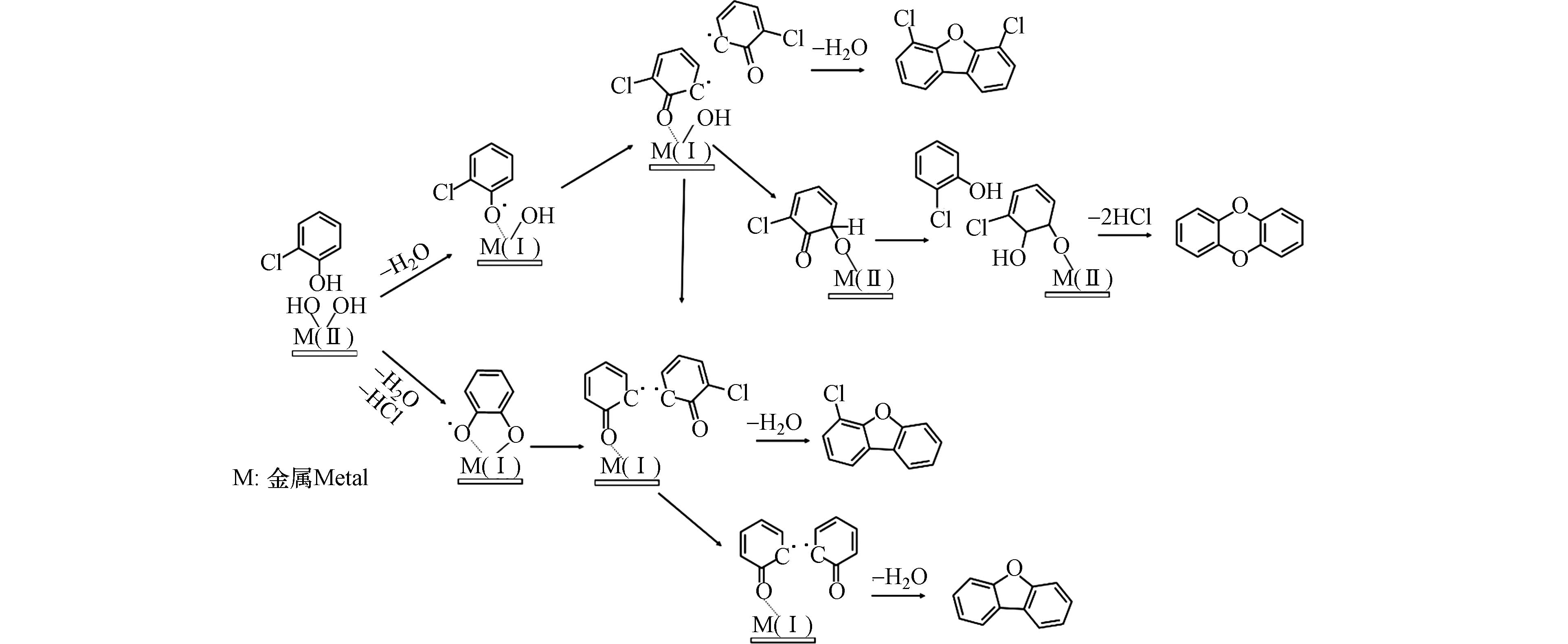
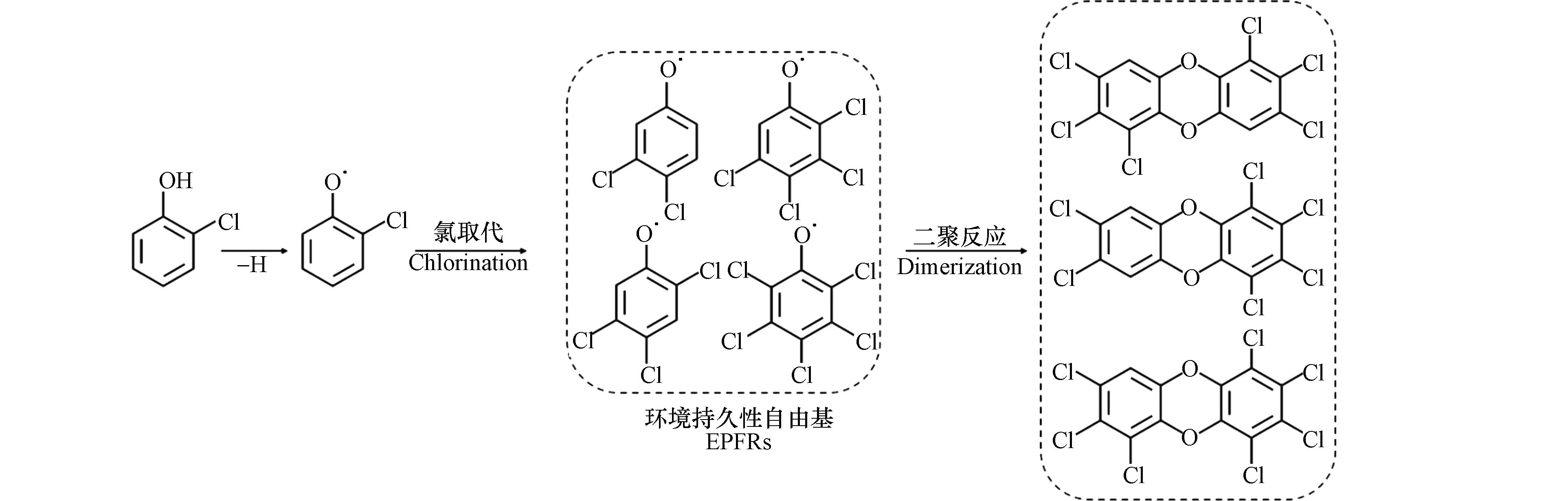
表 1 不同工况下、不同前驱体在不同金属氧化物上生成的EPFRs和UPOPs
Table 1. Summary of EPFRs and UPOPs generated from various precursors on different metal oxides under different conditions
前驱体
Precursor反应温度
Temperature反应气氛
Reaction atmosphere金属氧化物
Metal oxideEPFRs类型
Types of EPFRsUPOPs 参考文献
References对苯二酚、邻苯二酚、
苯酚400—800 °C 惰性 CuO 苯氧自由基、半醌自由基、环戊二烯自由基 N.A. [20] 对苯二酚、邻苯二酚、
苯酚、氯酚、一氯苯、
1,2-二氯苯100—400 °C 真空 CuO、Fe2O3、NiO、ZnO 苯氧自由基、半醌自由基 N.A. [29 − 32] 苯 230 °C 真空、惰性 CuxO 苯氧自由基 N.A. [33] 蒽、芘、苯并[a]芘、
9,10-蒽醌、1,4-萘醌、
苯并[g,h,i]苝室温 O3 N.A. 半醌类自由基、碳中心的多环芳烃自由基 N.A. [34] 2,4-二氯-1-萘酚 25—300 °C 空气 CuO、Al2O3、ZnO、NiO 萘氧自由基、萘醌自由基 N.A. [35] 苯酚 室温和250 °C O2 α-Fe2O3 苯氧自由基 N.A. [36] 邻苯二酚 25—327 °C O2 CuO、Fe2O3、CaO 氧中心苯氧自由基、半醌自由基 N.A. [37] 苯酚 250 °C 真空 ZnO、CuO、Fe2O3、TiO2 氧中心和碳中心的苯氧自由基 N.A. [38] 苯酚、邻苯二酚(理论计算) 27—727 °C 真空 Cu和Fe及其氧化物 酚醛类和苯氧自由基 N.A. [39] 苯酚 室温和250 °C 真空 ZnO 苯氧自由基 N.A. [40] 苯酚(理论计算) 27—727 °C 真空 α-Al2O3 苯氧自由基 N.A. [41] 苯酚 232 °C 真空 Al2O3、TiO2 苯氧自由基、半醌自由基 N.A. [42] 一氯苯、苯酚、1,2-二氯苯 230 °C 真空 PbO 苯氧自由基、苯基自由基 N.A. [43] 一氯苯、2-氯酚、
1,2-二氯苯230 °C 真空 CuO 苯氧自由基、半醌自由基 N.A. [44] 2-氯酚 225—500 °C 惰性和氧化 CuO 碳中心苯氧自由基 DD、MCDD、DCDF [45 − 46] 1,2-二氯苯 200—550 °C 惰性和氧化 CuO 苯氧自由基、半醌自由基 DD、MCDD、DCDF、DF [47] 2-氯酚 200—550 °C O2 Fe2O3 苯氧自由基、半醌自由基 DD、DF、MCDD/F、DCDD/F、TrCDD [48] 2,3,6-三氯酚 25—250 °C O2 CuO 苯氧自由基 PCDD/Fs [49] 1,2,3-三氯苯 250—550 °C 不同浓度O2 CuO、Fe2O3 苯氧自由基、半醌自由基 DF、PCDD/Fs [50] 氯酚 600 °C 氧化气氛 N.A. 苯氧自由基、环戊二烯自由基 PCNs、PCDFs [5] N.A., 不适用. DD, 二苯并-对-二噁英. MCDD, 一氯代二苯并-对-二噁英. DCDF, 二氯代二苯并呋喃. DF, 二苯并呋喃. TrCDD, 三氯代二苯并-对-二噁英.  下载: 导出CSV
下载: 导出CSV
表 2 不同阻滞剂对UPOPs的阻滞机理
Table 2. Inhibition mechanisms of various inhibitors on UPOPs
阻滞剂类型
Inhibitor category常用阻滞剂
Commonly used inhibitor影响因素
Impact factor阻滞机理
Inhibition mechanism碱性阻滞剂 Na2CO3、CaO、CaCO3、Ca(OH)2 Ca/Cl比值、温度 ● 吸收氯源,抑制有机氯生成[59]
● 将金属氯化物转化为金属氧化物,降低催化活性[60]
● 分解前驱体[26]硫基阻滞剂 S、SO2、Na2S2O3 S/Cl比值、温度 ● 将催化剂转化为硫酸盐或金属硫化物,降低催化活性[61]
● 与Cl2反应,消耗氯源[62]
● 磺化前驱体[63]氮基阻滞剂 NH3、NH4NO3、尿素 N/Cl比值、温度、
添加量● 与Cl2反应,消耗氯源[64]
● 与前驱体反应,生成芳香胺、氰化物和吡啶类化合物[65]
● 与金属催化剂螯合生成稳定络合物[66]
下载: 导出CSV
-
[1] LIU G R, ZHENG M H, CAI M W, et al. Atmospheric emission of polychlorinated biphenyls from multiple industrial thermal processes[J]. Chemosphere, 2013, 90(9): 2453-2460. doi: 10.1016/j.chemosphere.2012.11.008 [2] LIU G R, CAI Z W, ZHENG M H. Sources of unintentionally produced polychlorinated naphthalenes[J]. Chemosphere, 2014, 94: 1-12. doi: 10.1016/j.chemosphere.2013.09.021 [3] 王得梁, 谢雯静, 赵文博, 等. 工业过程二恶英的排放特征及其控制技术[J]. 环境化学, 2023, 42(5): 1449-1465. doi: 10.7524/j.issn.0254-6108.2022070704 WANG D L, XIE W J, ZHAO W B, et al. Dioxin emission characteristics and control technologies in industrial processes[J]. Environmental Chemistry, 2023, 42(5): 1449-1465 (in Chinese). doi: 10.7524/j.issn.0254-6108.2022070704
[4] YANG Y P, WU G L, JIANG C, et al. Variations of PCDD/Fs emissions from secondary nonferrous smelting plants and towards to their source emission reduction[J]. Environmental Pollution, 2020, 260: 113946. doi: 10.1016/j.envpol.2020.113946 [5] KIM D H, MULHOLLAND J A, RYU J Y. Formation of polychlorinated naphthalenes from chlorophenols[J]. Proceedings of the Combustion Institute, 2004, 30(1): 1245-1253. [6] STANMORE B R. The formation of dioxins in combustion systems[J]. Combustion and Flame, 2004, 136(3): 398-427. doi: 10.1016/j.combustflame.2003.11.004 [7] WEHRMEIER A, LENOIR D, SIDHU S S, et al. Role of copper species in chlorination and condensation reactions of acetylene[J]. Environmental Science & Technology:ES& T, 1998, 32(18): 2741-2748. [8] TAYLOR P H, SIDHU S S, RUBEY W A, et al. Evidence for a unified pathway of dioxin formation from aliphatic hydrocarbons[J]. Symposium (International) on Combustion, 1998, 27(2): 1769-1775. doi: 10.1016/S0082-0784(98)80018-0 [9] RYU J Y, MULHOLLAND J A. Metal-mediated chlorinated dibenzo-p-dioxin (CDD) and dibenzofuran (CDF) formation from phenols[J]. Chemosphere, 2005, 58(7): 977-988. doi: 10.1016/j.chemosphere.2004.08.084 [10] WEBER R, LINO F, IMAGAWA T, et al. Formation of PCDF, PCDD, PCB, and PCN in de novo synthesis from PAH: Mechanistic aspects and correlation to fluidized bed incinerators[J]. Chemosphere, 2001, 44(6): 1429-1438. doi: 10.1016/S0045-6535(00)00508-7 [11] WANG W, MA Y, LI S Y, et al. Effect of temperature on the EPR properties of oil shale pyrolysates[J]. Energy & Fuels, 2016: acs. energyfuels. 5b02211. [12] ARUOMA O I. Free radicals, oxidative stress, and antioxidants in human health and disease[J]. Journal of the American Oil Chemists’ Society, 1998, 75(2): 199-212. doi: 10.1007/s11746-998-0032-9 [13] CHEN Q C, SUN H Y, MU Z, et al. Characteristics of environmentally persistent free radicals in PM2.5: Concentrations, species and sources in Xi’an, Northwestern China[J]. Environmental Pollution, 2019, 247: 18-26. doi: 10.1016/j.envpol.2019.01.015 [14] SIGMUND G, SANTÍN C, PIGNITTER M, et al. Environmentally persistent free radicals are ubiquitous in wildfire charcoals and remain stable for years[J]. Communications Earth & Environment, 2021, 2: 68. [15] FENG W L, ZHANG Y F, HUANG L L, et al. Spatial distribution, pollution characterization, and risk assessment of environmentally persistent free radicals in urban road dust from central China[J]. Environmental Pollution, 2022, 298: 118861. doi: 10.1016/j.envpol.2022.118861 [16] SQUADRITO G L, CUETO R, DELLINGER B, et al. Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter[J]. Free Radical Biology & Medicine, 2001, 31(9): 1132-1138. [17] LIAO S H, PAN B, LI H, et al. Detecting free radicals in biochars and determining their ability to inhibit the germination and growth of corn, wheat and rice seedlings[J]. Environmental Science & Technology, 2014, 48(15): 8581-8587. [18] VEJERANO E P, RAO G Y, KHACHATRYAN L, et al. Environmentally persistent free radicals: Insights on a new class of pollutants[J]. Environmental Science & Technology, 2018, 52(5): 2468-2481. [19] ZHAO S, GAO P, MIAO D, et al. Formation and evolution of solvent-extracted and nonextractable environmentally persistent free radicals in fly ash of municipal solid waste incinerators[J]. Environmental Science & Technology, 2019, 53(17): 10120-10130. [20] DELLINGER B, LOMNICKI S, KHACHATRYAN L, et al. Formation and stabilization of persistent free radicals[J]. Proceedings of the Combustion Institute. International Symposium on Combustion, 2007, 31(1): 521-528. [21] LIU X Y, YANG L L, LIU G R, et al. Formation of environmentally persistent free radicals during thermochemical processes and their correlations with unintentional persistent organic pollutants[J]. Environmental Science & Technology, 2021, 55(10): 6529-6541. [22] IINO F, IMAGAWA T, TAKEUCHI M, et al. Formation rates of polychlorinated dibenzofurans and dibenzo-p-dioxins from polycyclic aromatic hydrocarbons, activated carbon and phenol[J]. Chemosphere, 1999, 39(15): 2749-2756. doi: 10.1016/S0045-6535(99)00209-X [23] ZHANG M M, YANG J, BUEKENS A, et al. PCDD/F catalysis by metal chlorides and oxides[J]. Chemosphere, 2016, 159: 536-544. doi: 10.1016/j.chemosphere.2016.06.049 [24] FUJIMORI T, TAKAOKA M, TAKEDA N. Influence of Cu, Fe, Pb, and Zn chlorides and oxides on formation of chlorinated aromatic compounds in MSWI fly ash[J]. Environmental Science & Technology, 2009, 43(21): 8053-8059. [25] FUJIMORI T, NAKAMURA M, TAKAOKA M, et al. Synergetic inhibition of thermochemical formation of chlorinated aromatics by sulfur and nitrogen derived from thiourea: Multielement characterizations[J]. Journal of Hazardous Materials, 2016, 311: 43-50. doi: 10.1016/j.jhazmat.2016.02.054 [26] LI Q Q, LI L W, SU G J, et al. Synergetic inhibition of PCDD/F formation from pentachlorophenol by mixtures of urea and calcium oxide[J]. Journal of Hazardous Materials, 2016, 317: 394-402. doi: 10.1016/j.jhazmat.2016.05.090 [27] DELHAES P, MARCHAND A. Analyse de la forme et de la position de signaix rpe observes sur des carbones graphitiques pulverulents[J]. Carbon, 1968, 6(2): 257-266. doi: 10.1016/0008-6223(68)90493-4 [28] HALES B J. Immobilized radicals. I. Principal electron spin resonance parameters of the benzosemiquinone radical[J]. Journal of the American Chemical Society, 1975, 97(21): 5993-5997. doi: 10.1021/ja00854a007 [29] LOMNICKI S, TRUONG H, VEJERANO E, et al. Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter[J]. Environmental Science & Technology, 2008, 42(13): 4982-4988. [30] VEJERANO E, LOMNICKI S, DELLINGER B. Formation and stabilization of combustion-generated environmentally persistent free radicals on an Fe(III)2O3/silica surface[J]. Environmental Science & Technology, 2011, 45(2): 589-594. [31] VEJERANO E, LOMNICKI S M, DELLINGER B. Formation and stabilization of combustion-generated, environmentally persistent radicals on Ni(II)O supported on a silica surface[J]. Environmental Science & Technology, 2012, 46(17): 9406-9411. [32] VEJERANO E, LOMNICKI S, DELLINGER B. Lifetime of combustion-generated environmentally persistent free radicals on Zn(II)O and other transition metal oxides[J]. Journal of Environmental Monitoring:JEM, 2012, 14(10): 2803-2806. doi: 10.1039/c2em30545c [33] D’ARIENZO M, GAMBA L, MORAZZONI F, et al. Experimental and theoretical investigation on the catalytic generation of environmentally persistent free radicals from benzene[J]. The Journal of Physical Chemistry C, 2017, 121(17): 9381-9393. doi: 10.1021/acs.jpcc.7b01449 [34] BORROWMAN C K, ZHOU S M, BURROW T E, et al. Formation of environmentally persistent free radicals from the heterogeneous reaction of ozone and polycyclic aromatic compounds[J]. Physical Chemistry Chemical Physics:PCCP, 2016, 18(1): 205-212. doi: 10.1039/C5CP05606C [35] YANG L L, LIU G R, ZHENG M H, et al. Pivotal roles of metal oxides in the formation of environmentally persistent free radicals[J]. Environmental Science & Technology, 2017, 51(21): 12329-12336. [36] SAKR N I, KIZILKAYA O, CARLSON S F, et al. Formation of environmentally persistent free radicals (EPFRs) on the phenol-dosed α-Fe2O3(0001) surface[J]. The Journal of Physical Chemistry. C, Nanomaterials and Interfaces, 2021, 125(40): 21882-21890. doi: 10.1021/acs.jpcc.1c04298 [37] QIN L J, YANG L L, LIU X Y, et al. Formation of environmentally persistent free radicals from thermochemical reactions of catechol[J]. The Science of the Total Environment, 2021, 772: 145313. doi: 10.1016/j.scitotenv.2021.145313 [38] SAKR N I, PATTERSON M C, DAEMEN L, et al. Vibrational and structural studies of environmentally persistent free radicals formed by phenol-dosed metal oxide nanoparticles[J]. Langmuir:the ACS Journal of Surfaces and Colloids, 2019, 35(51): 16726-16733. doi: 10.1021/acs.langmuir.9b02948 [39] AHMED O H, ALTARAWNEH M, AL-HARAHSHEH M, et al. Formation of phenoxy-type Environmental Persistent Free Radicals (EPFRs) from dissociative adsorption of phenol on Cu/Fe and their partial oxides[J]. Chemosphere, 2020, 240: 124921. doi: 10.1016/j.chemosphere.2019.124921 [40] PATTERSON M C, DiTUSA M F, McFERRIN C A, et al. Formation of environmentally persistent free radicals (EPFRs) on ZnO at room temperature: Implications for the fundamental model of EPFR generation[J]. Chemical Physics Letters, 2017, 670: 5-10. doi: 10.1016/j.cplett.2016.12.061 [41] ASSAF N W, ALTARAWNEH M, OLUWOYE I, et al. Formation of environmentally persistent free radicals on α-Al2O3[J]. Environmental Science & Technology, 2016, 50(20): 11094-11102. [42] PATTERSON M C, KEILBART N D, KIRURI L W, et al. EPFR Formation from Phenol adsorption on Al2O3 and TiO2: EPR and EELS studies[J]. Chemical Physics, 2013, 422: 277-282. doi: 10.1016/j.chemphys.2012.12.003 [43] WU J Z, LIU Y, ZHANG J, et al. A density functional theory calculation for revealing environmentally persistent free radicals generated on PbO particulate[J]. Chemosphere, 2020, 255: 126910. doi: 10.1016/j.chemosphere.2020.126910 [44] KIRURI L W, KHACHATRYAN L, DELLINGER B, et al. Effect of copper oxide concentration on the formation and persistency of environmentally persistent free radicals (EPFRs) in particulates[J]. Environmental Science & Technology, 2014, 48(4): 2212-2217. [45] LOMNICKI S, DELLINGER B. A detailed mechanism of the surface-mediated formation of PCDD/F from the oxidation of 2-chlorophenol on a CuO/silica surface[J]. The Journal of Physical Chemistry A, 2003, 107(22): 4387-4395. doi: 10.1021/jp026045z [46] LOMNICKI S, DELLINGER B. Formation of PCDD/F from the pyrolysis of 2-chlorophenol on the surface of dispersed copper oxide particles[J]. Proceedings of the Combustion Institute, 2002, 29(2): 2463-2468. doi: 10.1016/S1540-7489(02)80300-5 [47] NGANAI S, LOMNICKI S M, DELLINGER B. Formation of PCDD/Fs from the copper oxide-mediated pyrolysis and oxidation of 1, 2-dichlorobenzene[J]. Environmental Science & Technology, 2011, 45(3): 1034-1040. [48] GUAN X, GHIMIRE A, POTTER P M, et al. Role of Fe2O3 in fly ash surrogate on PCDD/Fs formation from 2-monochlorophenol[J]. Chemosphere, 2019, 226: 809-816. doi: 10.1016/j.chemosphere.2019.03.175 [49] YANG L L, LIU G R, ZHENG M H, et al. Molecular Mechanism of Dioxin Formation from Chlorophenol based on Electron Paramagnetic Resonance Spectroscopy[J]. Environmental Science & Technology, 2017, 51(9): 4999-5007. [50] CHEN T, SUN C, WANG T J, et al. Formation of DF, PCDD/Fs and EPFRs from 1, 2, 3-trichlorobenzene over metal oxide/silica surface[J]. Waste Management, 2020, 118: 27-35. doi: 10.1016/j.wasman.2020.08.024 [51] LI Z H, KONG B, WEI A Z, et al. Free radical reaction characteristics of coal low-temperature oxidation and its inhibition method[J]. Environmental Science and Pollution Research, 2016, 23(23): 23593-23605. doi: 10.1007/s11356-016-7589-x [52] KHACHATRYAN L, LOMNICKI S, DELLINGER B. An expanded reaction kinetic model of the CuO surface-mediated formation of PCDD/F from pyrolysis of 2-chlorophenol[J]. Chemosphere, 2007, 68(9): 1741-1750. doi: 10.1016/j.chemosphere.2007.03.042 [53] ALSOUFI A, ALTARAWNEH M, DLUGOGORSKI B Z, et al. A DFT study on the self-coupling reactions of the three isomeric semiquinone radicals[J]. Journal of Molecular Structure:THEOCHEM, 2010, 958(1/2/3): 106-115. [54] LIU X Y, LIU G R, LIU S T, et al. Free radical mechanism of toxic organic compound formations from o-chlorophenol[J]. Journal of Hazardous Materials, 2023, 443(Pt B): 130367. [55] SCHOONENBOOM M H, OLIE K. Formation of PCDDs and PCDFs from anthracene and chloroanthracene in a model fly ash system[J]. Environmental Science & Technology, 1995, 29(8): 2005-2009. [56] LIN B C, YANG L L, ZHENG M H, et al. Synergetic promoting/inhibiting mechanisms of copper/calcium compounds in the formation of persistent organic pollutants and environmentally persistent free radicals from anthracene[J]. Chemical Engineering Journal, 2022, 441: 136102. doi: 10.1016/j.cej.2022.136102 [57] MA Y F, WANG P Y, LIN X Q, et al. Formation and inhibition of Polychlorinated-ρ-dibenzodioxins and dibenzofurans from mechanical grate municipal solid waste incineration systems[J]. Journal of Hazardous Materials, 2021, 403: 123812. doi: 10.1016/j.jhazmat.2020.123812 [58] 徐帅玺. 典型钢铁生产过程二恶英生成机理及抑制研究[D]. 杭州: 浙江大学, 2018. XU S X. Study on mechanism of PCDD/fs formation and inhibition during steel manufacture process[D]. Hangzhou: Zhejiang University, 2018 (in Chinese).
[59] TSUYUMOTO I, KINOMURA M, KUZUHARA K. Inhibition of dioxin formation in flue gas by removal of hydrogen chloride using foaming water glass[J]. Journal of the Ceramic Society of Japan, 2006, 114(1329): 408-410. doi: 10.2109/jcersj.114.408 [60] ZHANG H, LAN D Y, LÜ F, et al. Inhibition of chlorobenzenes formation by calcium oxide during solid waste incineration[J]. Journal of Hazardous Materials, 2020, 400: 123321. doi: 10.1016/j.jhazmat.2020.123321 [61] RYAN S P, LI X D, GULLETT B K, et al. Experimental study on the effect of SO2 on PCDD/F emissions: Determination of the importance of gas-phase versus solid-phase reactions in PCDD/F formation[J]. Environmental Science & Technology, 2006, 40(22): 7040-7047. [62] RAGHUNATHAN K, GULLETT B K. Role of sulfur in reducing PCDD and PCDF formation[J]. Environmental Science & Technology, 1996, 30(6): 1827-1834. [63] TUPPURAINEN K, HALONEN I, RUOKOJÄRVI P, et al. Formation of PCDDs and PCDFs in municipal waste incineration and its inhibition mechanisms: A review[J]. Chemosphere, 1998, 36(7): 1493-1511. doi: 10.1016/S0045-6535(97)10048-0 [64] 龙红明, 李家新, 王平, 等. 尿素对减少铁矿烧结过程二恶英排放的作用机理[J]. 过程工程学报, 2010, 10(5): 944-949. LONG H M, LI J X, WANG P, et al. Reaction mechanism of emission reduction of dioxin by addition of urea in iron ore sintering process[J]. The Chinese Journal of Process Engineering, 2010, 10(5): 944-949 (in Chinese).
[65] KUZUHARA S, SATO H, TSUBOUCHI N, et al. Effect of nitrogen-containing compounds on polychlorinated dibenzo-p-dioxin/dibenzofuran formation through de novo synthesis[J]. Environmental Science & Technology, 2005, 39(3): 795-799. [66] LUNA A, AMEKRAZ B, MORIZUR J P, et al. Reactions of urea with Cu+ in the gas phase: an experimental and theoretical study[J]. The Journal of Physical Chemistry A, 2000, 104(14): 3132-3141. doi: 10.1021/jp9934634 [67] 钱永, 郑明辉, 刘文彬, 等. 钙化合物对前生体生成二噁英类的阻滞作用[J]. 环境化学, 2005, 24(6): 633-637. doi: 10.3321/j.issn:0254-6108.2005.06.002 QIAN Y, ZHENG M H, LIU W B, et al. Inhibition of calcium oxide, hydroxide and salts on pcdd/fs formation from precursors[J]. Environmental Chemistry, 2005, 24(6): 633-637 (in Chinese). doi: 10.3321/j.issn:0254-6108.2005.06.002
[68] WANG Y F, WANG L C, HSIEH L T, et al. Effect of temperature and CaO addition on the removal of polychlorinated dibenzo-p-dioxins and dibenzofurans in fly ash from a medical waste incinerator[J]. Aerosol and Air Quality Research, 2012, 12(2): 191-199. doi: 10.4209/aaqr.2011.06.0079 [69] MA H T, DU N, LIN X Y, et al. Inhibition of element sulfur and calcium oxide on the formation of PCDD/Fs during co-combustion experiment of municipal solid waste[J]. The Science of the Total Environment, 2018, 633: 1263-1271. doi: 10.1016/j.scitotenv.2018.03.282 [70] LUNDIN L, MOLTÓ J, FULLANA A. Low temperature thermal degradation of PCDD/Fs in soil using nanosized particles of zerovalent iron and CaO[J]. Chemosphere, 2013, 91(6): 740-744. doi: 10.1016/j.chemosphere.2013.02.021 [71] LIU W B, ZHENG M H, ZHANG B, et al. Inhibition of PCDD/Fs formation from dioxin precursors by calcium oxide[J]. Chemosphere, 2005, 60(6): 785-790. doi: 10.1016/j.chemosphere.2005.04.020 [72] LIN X Q, JI L J, ZHAN M X, et al. Suppression of PCDD/fs by raw meal in cement kilns[J]. Aerosol and Air Quality Research, 2018, 18(4): 1032-1043. doi: 10.4209/aaqr.2018.01.0023 [73] YAN J H, PENG Z, LU S Y, et al. Degradation of PCDD/Fs by mechanochemical treatment of fly ash from medical waste incineration[J]. Journal of Hazardous Materials, 2007, 147(1/2): 652-657. [74] 卫樱蕾, 严建华, 陆胜勇, 等. 钙基添加剂对机械化学法降解二恶英的影响[J]. 浙江大学学报(工学版), 2010, 44(5): 991-997. doi: 10.3785/j.issn.1008-973X.2010.05.026 WEI Y L, YAN J H, LU S Y, et al. Decomposition of PCDD/Fs by mechanochemical means with calcium-based additives[J]. Journal of Zhejiang University (Engineering Science), 2010, 44(5): 991-997 (in Chinese). doi: 10.3785/j.issn.1008-973X.2010.05.026
[75] WANG X X, LV J B, YING Y X, et al. A new insight into the CaO-induced inhibition pathways on PCDD/F formation: Metal passivation, dechlorination and hydroxide substitution[J]. The Science of the Total Environment, 2023, 885: 163782. doi: 10.1016/j.scitotenv.2023.163782 [76] SHAO K, YAN J H, LI X D, et al. Inhibition of de novo synthesis of PCDD/Fs by SO2 in a model system[J]. Chemosphere, 2010, 78(10): 1230-1235. doi: 10.1016/j.chemosphere.2009.12.043 [77] LONG H M, LI J X, WANG P, et al. Emission reduction of dioxin in iron ore sintering by adding urea as inhibitor[J]. Ironmaking & Steelmaking, 2011, 38(4): 258-262. [78] WANG P J, XIE F, YAN F, et al. Inhibitory effect and mechanism of an N-S-based inhibitor (CH4N2S) on PCDD/fs in flue gas and fly ash in a full-scale municipal solid waste incinerator[J]. ACS ES& T Engineering, 2023, 3(10): 1557-1567. [79] ZHANG Y D, BUEKENS A, LIU L N, et al. Suppression of chlorinated aromatics by nitrogen and sulphur inhibitors in iron ore sintering[J]. Chemosphere, 2016, 155: 300-307. doi: 10.1016/j.chemosphere.2016.04.065 [80] MA Y F, LIN X Q, CHEN Z L, et al. Influences of P-N-containing inhibitor and memory effect on PCDD/F emissions during the full-scale municipal solid waste incineration[J]. Chemosphere, 2019, 228: 495-502. doi: 10.1016/j.chemosphere.2019.04.161 [81] HUNSINGER H, SEIFERT H, JAY K. Reduction of PCDD/F formation in MSWI by a process-integrated SO2 cycle[J]. Environmental Engineering Science, 2007, 24(8): 1145-1159. doi: 10.1089/ees.2007.0108 [82] 付建英, 陈彤, 吴海龙, 等. SO2抑制二噁英从头合成的实验及其过程模拟[J]. 化工学报, 2014, 65(9): 3687-3693. FU J Y, CHEN T, WU H L, et al. Experiments and reaction simulation for SO2 inhibition on de novo formation of PCDD/Fs[J]. CIESC Journal, 2014, 65(9): 3687-3693 (in Chinese).
[83] 杨帆, 黎烈武, 童东革, 等. 钙基复合氧化物对五氯酚生成二噁英的阻滞作用[J]. 环境化学, 2015, 34(8): 1439-1445. doi: 10.7524/j.issn.0254-6108.2015.08.2015013002 YANG F, LI L W, TONG D G, et al. Inhibition of calcium-based composite oxide on PCDD/Fs formation from PCP[J]. Environmental Chemistry, 2015, 34(8): 1439-1445 (in Chinese). doi: 10.7524/j.issn.0254-6108.2015.08.2015013002
-
 点击查看大图
点击查看大图
计量
- 文章访问数: 1052
- HTML全文浏览数: 1052
- PDF下载数: 22
- 施引文献: 0
