

-
在饮用水和污水处理系统中,往往采用氯化处理或者紫外辐射来灭活病原体以及控制介水疾病的爆发[1 − 2]. 水中的天然有机质或者污染物在此过程中不可避免地发生转化,生成消毒副产物,对水环境和人体健康构成潜在威胁[3]. 因此,近年发展出多种基于紫外光的水处理高级氧化技术,例如紫外/过氧化氢、紫外/臭氧、紫外/氯等,以期提高污染物去除效率,降低消毒副产物的生成[4]. 其中紫外/氯体系是将含氯水体经紫外辐射产生·OH和·Cl,而后经过自由基链传递反应生成·ClO、·Cl2−、·O−、·HOCl−等多种活性反应物种. 这些不同的活性自由基在紫外/氯体系中能够有效地降解水中的各种污染物. 相比于中性条件,在酸性条件(pH < 4.0)时,紫外/氯体系中产生的·OH和·Cl等活性自由基浓度相对更高,且活性氯自由基占比更大,对化合物的降解效果更好[5 − 6]. 碱性条件下的紫外/氯体系,·ClO的占比提升,而·OH和·Cl明显减少[7]. 因此,研究这些活性自由基在紫外/氯体系中的作用机制是十分必要的.
在紫外/氯体系的实验研究中,大多采用探针物质如硝基苯、对氯苯甲酸等,基于自由基的基元反应来计算目标物与各自由基的二级反应速率常数[8 − 9]. 其中,硝基苯和对氯苯甲酸由于与其他自由基反应活性较低,常用来当作·OH的探针物质[7,10 − 11];而由于苯甲酸与硝基苯在紫外/氯体系中反应速率的不同,通常采用这两种物质联用来作为·Cl的探针物质[12 − 13]. Lei等研究测定了多种探针化合物和·OH、·SO4−以及·Cl等活性物种之间的反应动力学数据,提出了探针化合物的准确性与淬灭剂剂量之间的联系[14]. 此外,在基于自由基稳态浓度的假设前提下,根据不同的基元反应方程,可以建立不同的数学模型来预测紫外/氯体系中的反应动力学方程[15 − 18]. Minakata等以线性自由能关系为基础,可以模拟水体中紫外/氯体系产生的多种无机自由基的反应活性,研究表明加成与抽氢是主要的反应方式[19]. 作为紫外/氯体系的研究基础,探针化合物与活性自由基之间的反应更为复杂,需要进一步明确其反应机理,从而可以更准确地确定探针化合物的使用范围.
环境理论化学是近年来快速发展的交叉学科. 量子化学基于分子中原子的电子波函数进行计算,可以模拟优化化合物的分子状态,分析其结构信息和电荷分布等基本性质,从而揭示化合物的物理化学性质并预测其环境行为. 例如通过理论计算化学,可以解释有机污染物在大气环境中的自由基反应、水环境中的水解反应等反应机理[20 − 23]. 通过非周期性体系问题的计算,明确污染物受活性物种(自由基、氧化剂和还原剂等)攻击的反应机理[24]. 因此,本文采取量子化学计算方法,研究了硝基苯、苯甲酸和对氯苯甲酸与紫外/氯体系中生成的3种主要自由基·OH、·ClO和·Cl的反应机制,以期为紫外/氯高级氧化技术的实际应用研究提供理论基础.
-
采用Gaussian16软件计算硝基苯(NB)、苯甲酸(BA)和对氯苯甲酸(pCBA)分子的几何构型和电子能量[25]. 反应物、过渡态以及中间体的构型优化和频率分析均在M06-2X/6-311+G(d,p)方法下进行计算,其中过渡态处于势能面鞍点且拥有唯一虚频,反应物及中间体均处于势能面驻点且振动频率皆为实频[26]. 在同一方法下,利用内禀反应坐标法计算了反应最低能量路径以确保每个过渡态均连接了正确的反应物与中间体. 利用优化好的构型在M06-2X/6-311+G(3df,2pd)方法下计算了分子单点能,并用M06-2X/6-311+G(d,p)方法下得到的零点能进行校正. 此外,选取了3种泛函B3LYP、M06-2X和ωB97进行测试来平衡计算精度与耗时之间的关系,最终确定研究体系所用的泛函为M06-2X.
考虑到实验环境为水溶液,采用隐式溶剂校正对体系进行了溶剂化效应的校正,选取基于密度的溶剂化模型(Solvation Model based on Density, SMD)校正化合物分子与水溶液之间的短程相互作用[27],该计算在M06-2X/6-311+G(3df,2pd)方法下完成.
-
反应速率常数基于过渡态理论计算,并采用Wigner系数校正了隧道效应,反应方程根据高斯的热力学定理推导[28],如式(1):
其中,σ是反应路径简并度,kB是玻尔兹曼常数,T是温度,h是普朗克常数,R是理想气体常数,P0是标准态大气压,
$ \mathrm{\Delta }n=n-1 $ ,n为参与反应的分子数,ΔG0,≠是反应的吉布斯自由能垒,其中,3种自由基(·OH,·ClO和·Cl)和3种化合物(NB+,BA和pCBA)发生加成反应与抽氢反应时,ΔG0,≠为过渡态与反应物之间的吉布斯自由能差. 3种自由基和3种化合物发生单电子转移反应时,则基于Marcus理论计算反应的ΔG0,≠ [29]. 计算如式(2)—(3).其中,
$ {\mathrm{\Delta }G}_{r}^{0} $ 是反应物与产物之间的吉布斯自由能变,$ \mathrm{\lambda } $ 是反应的重组能,包括内重组能($ {\lambda }_{i} $ )和外重组能($ {\lambda }_{0} $ ).κi是Wigner透射系数,计算如式(4):
其中,
$ {\nu }_{i}^{\ne } $ 是过渡态的虚频频率. -
首先测试了3种泛函B3LYP、M06-2X和ωB97对研究体系的影响,在M06-2X/6-311+G(d,p)下优化计算不同位点的反应路径,选取3种探针化合物(NB+、BA和pCBA)和3种自由基(·OH、·ClO、·Cl)之间的9种反应路径在6-311+G(3df,2pd)基组下进行泛函的测试,并与M06-2X/6-311+G(3df,2pd)计算优化结果进行对比,结果见表1,测试的9种反应路径,是在包含了所有反应类型后,随机选择的结果,其中包括了最低能垒反应通道,也包括了较高能垒的反应通道,以便在选择泛函时兼顾所有计算需求. 对比发现M06-2X的计算结果更接近M06-2X/6-311+G(3df,2pd)条件下的优化值,耗时较短. 且在研究有机化合物与自由基之间的反应时,M06-2X泛函精度较高,结果更可靠[30 − 31]. 因此选择M06-2X泛函进行后续反应机理的计算.
-
3种目标物的pKa值均在4左右,其形态分布图如图1所示,因此在自然水体环境下(pH = 6.0—8.0),硝基苯处于分子态,苯甲酸和对氯苯甲酸则是负离子态. 已研究了pH = 7.0条件下3种探针物质在紫外/氯体系中与自由基的反应机制[32]. 然而在酸性条件(pH < 4.0)时,紫外/氯体系中产生的·OH和·Cl等活性自由基的浓度相比中性条件更高,且活性氯自由基占比更大[5 − 7]. 而且硝基苯主要为正离子态,苯甲酸和对氯苯甲酸均为分子态. 因此,3种探针化合物在酸性条件下(pH < 4.0时)在紫外/氯体系中的降解情况可能会与中性条件下不同. 本文主要采用量子化学计算研究pH < 4.0时3种探针化合物与紫外/氯体系中3种主要自由基·OH、·ClO和·Cl的反应机制.
在M06-2X/6-311+G(3df,2pd)条件下优化计算硝基苯、苯甲酸和对氯苯甲酸的分子态及离子态构型,优化后的结构即该分子能量最低的稳定构型,如图2所示,(a)—(c)分别为硝基苯、苯甲酸和对氯苯甲酸的分子态构型,(d)—(f)分别为硝基苯、苯甲酸和对氯苯甲酸的离子态构型,以C1—C6对碳原子进行编号. 后续计算以图中优化后的构型为基础.
-
·OH是一种非选择性自由基,可与探针化合物的各个位点发生反应,包括加成反应和抽氢反应两种反应机制[33]. 离子态硝基苯与·OH加成反应(6条反应路径)与抽氢反应(5条反应路径)的反应能垒图见图3(a),因C2与C6、C3与C5为对称位点,能垒接近,因此图中只显示C1—C4位点的反应能垒. ·OH与离子态硝基苯、苯甲酸和对氯苯甲酸的吉布斯自由能垒(ΔG0,≠)、吉布斯自由能变(ΔG0)以及速率常数ki见表2. 离子态硝基苯与·OH的反应中,反应能垒显示各位点的抽氢反应均快于加成反应,在抽氢反应中,C1位点即对位反应能垒最低(7.7 kcal·mol−1),其次是邻位(C3(5),11.2 kcal·mol−1)和间位(C2(6),11.5 kcal·mol−1)反应能垒. 加成反应中,能垒最低的反应在邻位(C3(5),13.5 kcal·mol−1),对位反应能垒稍高(C1,16.9 kcal·mol−1),而间位以及C4位点的加成反应则能垒相对较高(19.2—20.1 kcal·mol−1). 苯甲酸与·OH的反应中,同样包括加成反应(6条反应路径)与抽氢反应(5条反应路径),见图3(b). 与离子态硝基苯类似,同样是抽氢反应快于加成反应,在抽氢反应中,邻位(C3(5),5.6 kcal·mol−1)反应能垒较低,对位(C1,6.4 kcal·mol−1)次之,间位(C2(6),7.4 kcal·mol−1)反应能垒略高,相比于抽氢反应,加成反应能垒普遍较高(7.7—10.0 kcal·mol−1). 对氯苯甲酸与·OH的反应见图3(c). 与离子态硝基苯和苯甲酸相似,同样是抽氢反应快于加成反应. 反应最快的位点是邻位(C3(5))的抽氢反应(5.2 kcal·mol−1),其次是间位(C2(6))的加成与抽氢反应(5.5—9.2 kcal·mol−1)及C4位点加成(9.4 kcal·mol−1),对位(C1)加成反应能垒最高(12.4 kcal·mol−1). 对比发现在与·OH的反应中,苯甲酸能垒最低,对氯苯甲酸次之,而离子态硝基苯最慢,可能是离子态硝基苯带正电荷,使自由基反应变慢.
-
·ClO也是紫外/氯体系中大量存在的自由基之一,相比于·OH,·ClO是一种选择性自由基,主要反应机制同样包括加成反应和抽氢反应. 分别计算了·ClO与离子态硝基苯、苯甲酸和对氯苯甲酸的反应通道. ·ClO与离子态硝基苯、苯甲酸和对氯苯甲酸的吉布斯自由能垒(ΔG0,≠)、吉布斯自由能变(ΔG0)以及速率常数ki见表3. 计算结果表明·ClO与3种化合物的反应能垒普遍高于·OH. 其中,离子态硝基苯与·ClO的反应能垒图见图4(a),与·OH反应不同,·ClO与离子态硝基苯的加成反应能垒低于抽氢反应,其中在·ClO加成反应中,邻位反应能垒最低(C3(5),12.2 kcal·mol−1),对位次之(C1,15.4 kcal·mol−1),间位(C2(6),20.9 kcal·mol−1)和C4位点(25.1 kcal·mol−1)较高,抽氢反应的能垒则略高,在22.1—24.7 kcal·mol−1范围. 苯甲酸与·ClO的反应能垒见图4(b),抽氢反应略快于加成反应. 其中,对位(C1)的抽氢反应为能量最低通道(11.8 kcal·mol−1),其余位点的加成反应和抽氢反应能垒差别不大(13.2—15.5 kcal·mol−1). 对氯苯甲酸与·ClO的反应能垒见图4(c),其中加成反应略快于抽氢反应,在加成反应中,间位加成能垒最低(C2(6),12.3 kcal·mol−1),与离子态硝基苯和苯甲酸不同的是,对位(C1)的加成反应能垒较高(15.6 kcal·mol−1),这可能是由于对位本身有氯取代基,使该位点与·ClO的反应变慢. 其余位点的反应能垒在13.3—17.6 kcal·mol−1范围. 对比发现,在与·ClO的反应中苯甲酸最快,对氯苯甲酸次之,离子态硝基苯最慢. 但是总体而言,与·ClO的反应能垒略高于与·OH的反应.
-
·Cl与污染物的反应主要以抽氢方式发生[21], Zhang等的研究也表明,·Cl难以加成在苯环中C—H处的C上[34]. ·OH与离子态硝基苯、苯甲酸和对氯苯甲酸的吉布斯自由能垒(ΔG0,≠)、吉布斯自由能变(ΔG0)以及速率常数ki见表4. 在离子态硝基苯与·Cl的反应中,间位(C2(6))是主要的反应通道,其反应能垒为10.3 kcal·mol−1,而其余位点则能垒较高,为14.1—15.7 kcal·mol−1,反应能垒见图5(a). 苯甲酸与·Cl的反应(图5(b)),则是对位(C1,5.4 kcal·mol−1)为主要的反应通道,其余位点反应能相比于离子态硝基苯略低,为10.3—11.9 kcal·mol−1. 对氯苯甲酸与·Cl的反应(图5(c)),只发生间位和邻位的抽氢反应,其中间位(C2(6),10.2 kcal·mol−1)快于邻位(C3(5),10.9 kcal·mol−1). 对比而言,苯甲酸与·Cl反应最快,离子态硝基苯和对氯苯甲酸则与·Cl反应较慢.
-
基于Marcus理论,根据式(2)—(3)分别计算了硝基苯、苯甲酸和对氯苯甲酸与3种自由基(·OH、·ClO和·Cl)单电子转移反应的自由能垒,结果见表5. 其中,离子态硝基苯与3种自由基的单电子转移能垒都较高,为10.8—17.6 kcal·mol−1,反应速率常数较小. 苯甲酸与3种自由基的单电子转移能垒则较低,其中与·Cl的单电子转移反应能垒最低,为5.8 kcal·mol−1. 对于对氯苯甲酸,·OH和·Cl的单电子转移能垒近似(8.2 kcal·mol−1),·ClO则能垒较高. 总体而言,·Cl更易引发单电子转移反应,·OH次之,·ClO则较难发生单电子转移反应.
-
基于反应中过渡态与反应物(复合形态)之间的能垒,利用过渡态理论和Marcus理论(式(1)—(4))计算了反应速率常数. 3种化合物与·OH的吉布斯自由能垒(ΔG0,≠)、吉布斯自由能变(ΔG0)以及速率常数ki见表2,与·ClO的结果见表3,与·Cl的结果见表4,3种化合物与3种自由基的反应速率常数ki见表5,根据式(5)—(7)分别计算每种探针化合物与3种自由基(·OH、·ClO和·Cl)的总反应速率常数
$ {k}_{\mathrm{C}\mathrm{P}-\mathrm{O}\mathrm{H}} $ 、$ {k}_{\mathrm{C}\mathrm{P}-\mathrm{C}\mathrm{l}\mathrm{O}} $ 和$ {k}_{\mathrm{C}\mathrm{P}-\mathrm{C}\mathrm{l}} $ ,其中CP表示化合物,包括硝基苯,苯甲酸和对氯苯甲酸,结果见表6.在pH < 4.0时,离子态硝基苯与·OH的反应是最快的,·ClO次之,·Cl最慢,这与实验检测的结果一致. 其中起主要作用的为·OH,而另外两种自由基则反应性较差[35]. 苯甲酸的反应顺序则是:·OH > ·Cl > ·ClO,其中·OH和·Cl 的反应速率在同一个数量级,·ClO的作用可忽略不计. 而在实验中,苯甲酸与·Cl的反应速率常数大于与·OH的反应速率常数[35],这可能是因为实验体系较为复杂,各自由基之间浓度不同,存在竞争关系,而计算的结果的精度受限于所选泛函和基组,因此与实验结果存在一定偏差. 对氯苯甲酸在紫外/氯体系的反应中,则是·OH占主导地位,·ClO和·Cl的反应速率远小于·OH. 对氯苯甲酸和硝基苯类似,都是作为·OH的探针物质被广泛应用到实验中[36]. 整体而言,在pH < 4.0的紫外/氯降解体系中,苯甲酸降解速率最快,对氯苯甲酸次之,硝基苯最慢. 而对比中性条件下的计算数据(表6),分子态硝基苯的反应速率远远大于离子态硝基苯,且·OH占主导作用. 对于苯甲酸,在中性条件下呈负离子态,与·OH的反应速率略有提升,与·Cl的反应速率大大降低,可能是在离子状态下,和·Cl只发生C3(5)位点的抽氢反应[32]. 离子态对氯苯甲酸则和3种自由基的反应均快于分子态,且都是·OH占主导地位. 总体而言,无论是酸性条件还是中性条件,与探针化合物反应最快的都是·OH,对于探针化合物的不同形态,其反应速率的顺序则是负离子态>分子态>正离子态.
-
本研究采用量子化学计算的方法研究了在酸性条件下,3种探针化合物硝基苯、苯甲酸和对氯苯甲酸与紫外/氯体系中的3种主要自由基·OH、·ClO和·Cl的反应机理,其中主要反应途径为加成反应、抽氢反应和单电子转移反应. 对于离子态硝基苯和对氯苯甲酸,起主要作用的是·OH,·ClO和·Cl占比相对较小,这与在中性条件下的反应机理相吻合. 对于苯甲酸, ·OH和·Cl与其反应速率在同一个数量级,其中·Cl的单电子转移反应贡献较高. 而对于不同形态的探针化合物,·OH作为非选择性自由基,和探针化合物的反应速率较快,而选择性自由基·ClO和·Cl的反应速率则与探针化合物形态有较大关系. 总体而言,对于探针化合物的不同形态,其反应速率的顺序为负离子态>分子态>正离子态.
基于理论化学计算研究探针物质在紫外/氯体系中的反应机理
Quantum chemical study on reaction mechanism of reactive radical mediated probe compounds degradation in the UV/chlorine process
-
摘要: 紫外/氯体系是将氯化消毒与紫外线辐射相结合的一种水处理高级氧化技术. 硝基苯、苯甲酸和对氯苯甲酸常被作为探针化合物,广泛用于研究紫外/氯体系中生成的羟基自由基(·OH)和活性氯自由基(·ClO和·Cl等). 采用理论化学计算的方法研究了硝基苯、苯甲酸和对氯苯甲酸与紫外/氯体系中生成的3种主要自由基·OH、·ClO和·Cl的反应机理. 在酸性条件下(pH <4.0),硝基苯、苯甲酸和对氯苯甲酸与·OH、·ClO和·Cl的反应包括抽氢反应、加成反应和单电子转移反应. ·OH与3种探针化合物反应的ΔG0,≠分别为7.7—20.1 kcal·mol−1、5.6—10.1 kcal·mol−1和5.2—12.4 kcal·mol−1. ·ClO与3种化合物的ΔG0,≠则相对较高为11.8—25.1 kcal·mol−1. ·Cl 引发的反应中,与硝基苯(10.3—14.1 kcal·mol−1)和对氯苯甲酸反应能垒较高(10.2—12.4 kcal·mol−1),与苯甲酸则反应能垒较低(5.4—11.9 kcal·mol−1),其中单电子转移反应起重要作用. 因此在紫外/氯体系中,起主要反应的是·OH,·ClO作用较小,·Cl的贡献则主要体现在抽氢反应和单电子转移反应中,这与在中性条件下的反应机理相吻合. 总体而言,对于探针化合物的不同形态,其反应速率的顺序为负离子态>分子态>正离子态. 本文的研究结果为探究紫外/氯体系当中各种自由基的反应机理提供了数据支撑.Abstract: The UV/chlorine process, by combining chlorination with UV irradiation, has been recently considered as a highly efficient advanced oxidation process in water treatment. Nitrobenzene (NB), benzoic acid (BA) and p-chlorobenzoic acid (pCBA) are widely used as model probe compounds in UV/chlorine system to investigate the second-order rate constants of specific radical-reaction with target contaminants by a competitive kinetics method. Here we evaluated the radical mediated reaction kinetics and mechanisms of NB, BA and pCBA in UV/chlorine process at pH<4.0 by theoretical calculations. The main reactive radicals ·OH, ·ClO and ·Cl in UV/chlorine process for the reaction with NB, BA and pCBA can be explained by H-abstraction, addition pathways and electron transfer reaction. The ΔG0,≠ values for ·OH reaction with NB, BA and pCBA were in the range of 7.7—20.1 kcal·mol−1, 5.6—10.1 kcal·mol−1 and 5.2—12.4 kcal·mol−1, respectively. The ΔG0,≠ values for ·ClO reactions with these three probe compounds were in the range of 11.8—25.1 kcal·mol−1, which were higher than those of ·OH. The ΔG0,≠ values for ·Cl reactions with NB and pCBA were in the range of 10.3—14.1 kcal·mol−1 and 10.2—12.4 kcal·mol−1. As for BA, the ΔG0,≠ values were in the range of 5.4—15.7 kcal·mol−1 due to the dominant role of the electron transfer reaction. This is consistent with the results under neutral conditions. For the different forms of probe compounds, the order of reaction rates is as follows: anionic form > molecular form > cationic form. The results of this study provided some basic data for investigations into the reaction kinetics and mechanisms of reactive radicals in UV/chlorine process.
-
Key words:
- UV/Chlorine /
- probe compounds /
- quantum chemistry.
-
-

图 1 (a)硝基苯、(b)苯甲酸和(c)对氯苯甲酸形态分布
Figure 1. The speciation of nitrobenzene (NB) (a), benzoic acid (BA) (b) and p-chlorobenzoic acid (pCBA) (c)
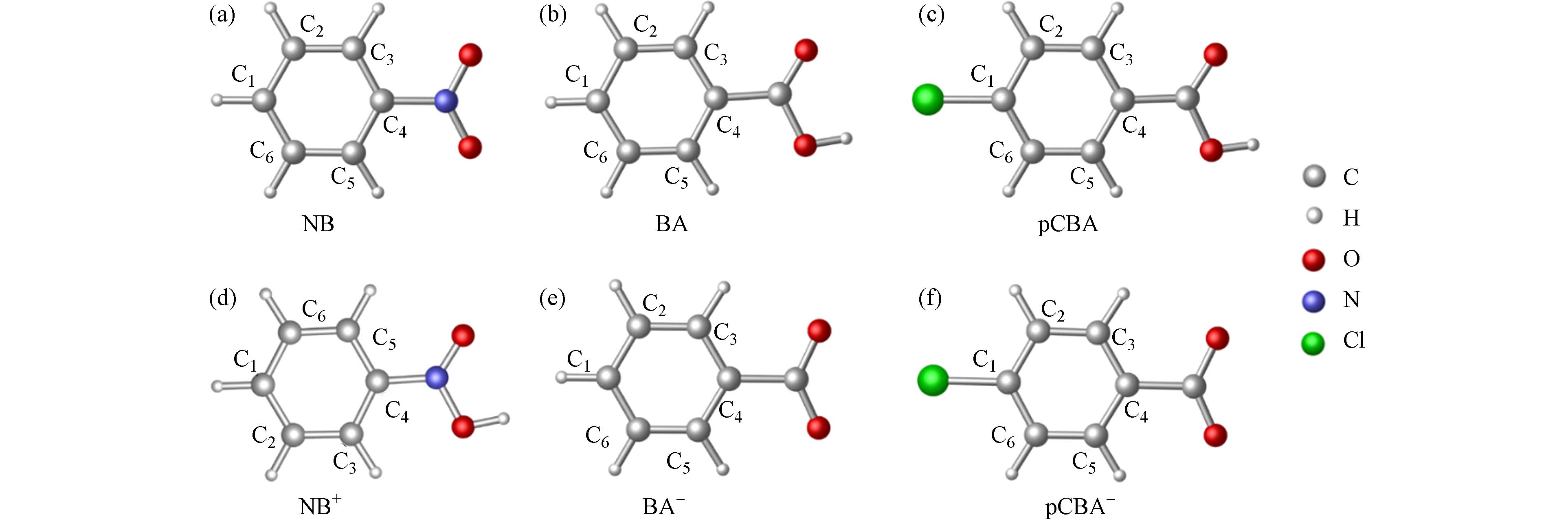
图 2 硝基苯、苯甲酸和对氯苯甲酸分子及离子态结构图(a)—(c)分别为硝基苯、苯甲酸和对氯苯甲酸的分子态构型,(d)—(f)分别为硝基苯、苯甲酸和对氯苯甲酸的离子态构型,以C1—C6对碳原子进行编号
Figure 2. The optimized molecular structure of nitrobenzene (NB), benzoic acid (BA) and p-chlorobenzoic acid (pCBA)

图 3 离子态硝基苯、苯甲酸以及对氯苯甲酸与·OH反应能垒图
Figure 3. Relative potential energies of reactions between NB+, BA, pCBA and ·OH (a) represents the reactions between NB+ and ·OH, (b) represents the reactions between BA and ·OH, (c) represents the reactions between pCBA and ·OH
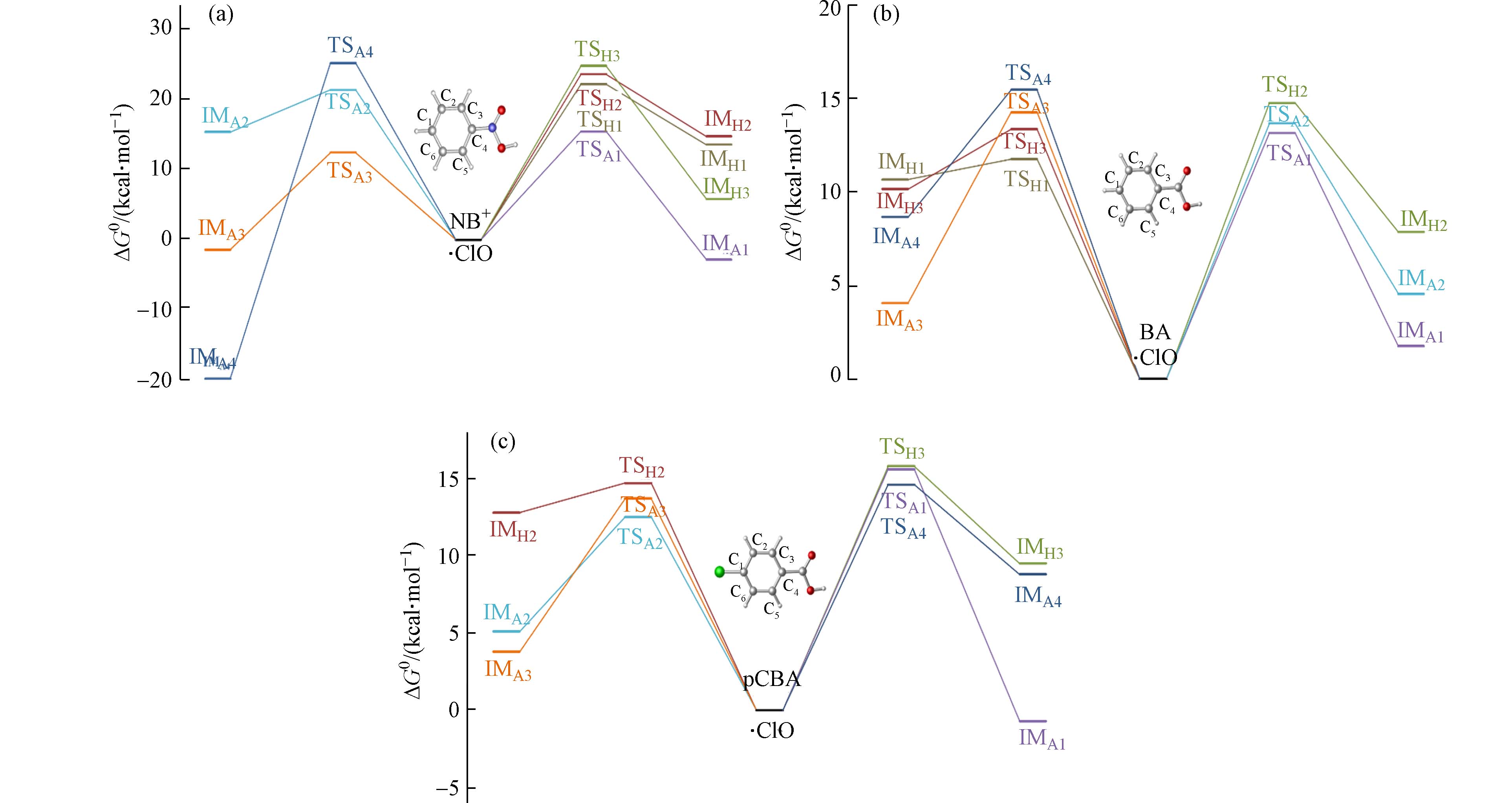
图 4 离子态硝基苯、苯甲酸以及对氯苯甲酸与·ClO反应能垒图
Figure 4. Relative potential energies of initial reactions between NB+, BA, pCBA and ·ClO
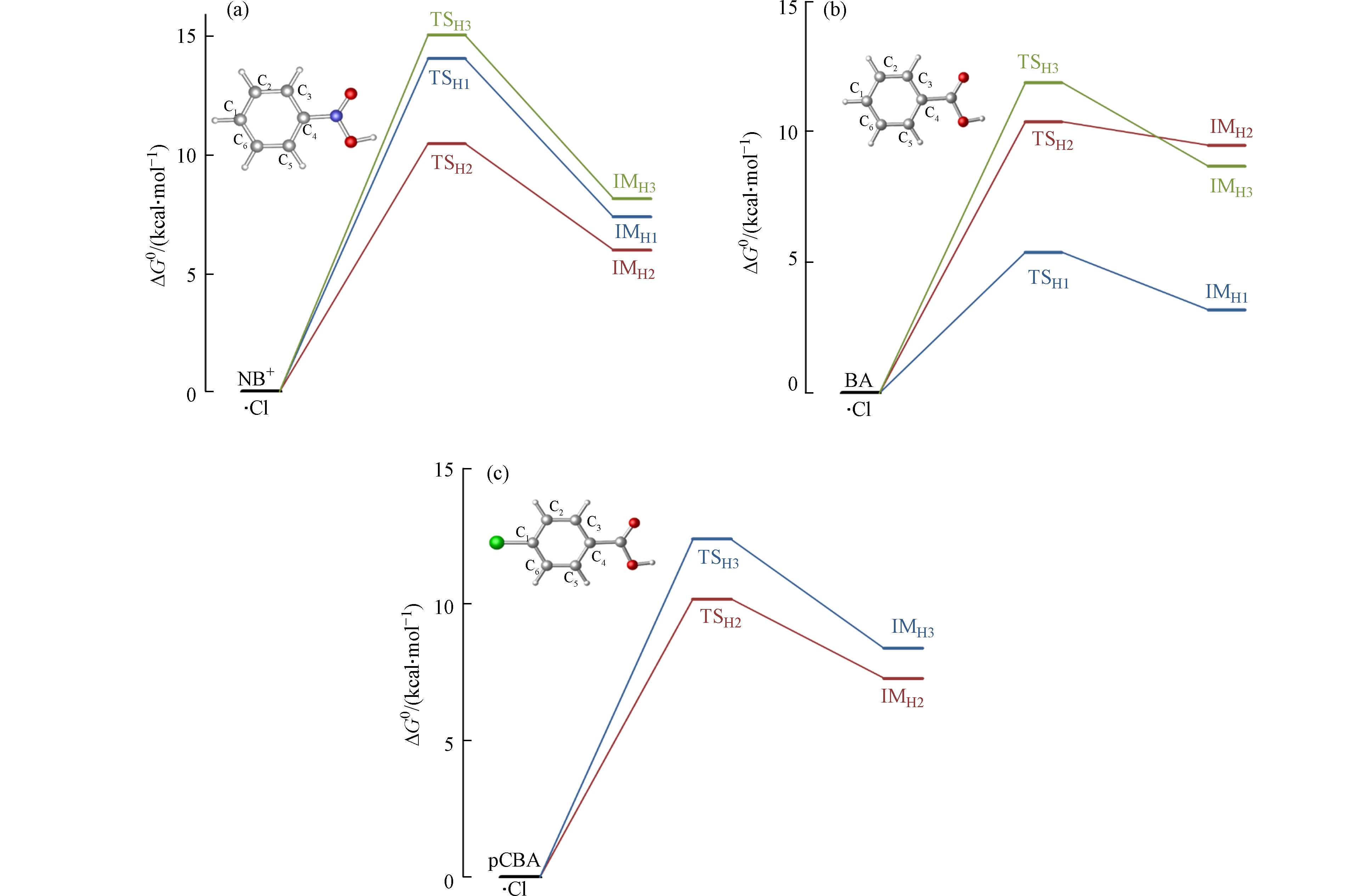
图 5 离子态硝基苯、苯甲酸以及对氯苯甲酸与·Cl反应能垒图
Figure 5. Relative potential energies of initial reactions between NB+, BA, pCBA and ·Cl
表 1 不同泛函计算相对势能(ΔE0,kcal·mol−1)与M06-2X/6-311+G(3df,2pd)计算优化结果对比
Table 1. Relative potential energy (ΔE0, kcal·mol−1) calculated by different functionals at 6-311+G(3df,2pd) level based on the optimized geometries from M06-2X/6-311+G(d,p) and compared with the results that optimized at M06-2X/6-311+G(3df,2pd) level
反应路径 1)
PathwaysB3LYP M06-2X ωB97 M06-2X opt B3LYP M06-2X ωB97 M06-2X opt ΔE0,≠/(kcal·mol−1) ΔE0/(kcal·mol−1) NB+_OH_C6_A 22.4 21.3 23.2 20.6 3.5 3.4 4.3 3.7 NB+_ClO_C3_A 10.6 12.2 12.5 12.2 −4.9 −1.5 −3.2 −2.5 NB+_Cl_C1_H 11.1 13.0 12.4 13.5 10.1 9.2 12.2 8.9 BA_OH_C1_A 5.7 4.9 6.2 4.7 −17.4 −13.8 −20.4 −13.1 BA_ClO_C1_A 13.5 10.7 14.1 9.7 −5.1 −2.6 −0.7 −1.6 BA_Cl_C3_H 19.9 16.4 20.4 17.2 10.2 9.8 10.8 9.1 pCBA_OH_C6_H 6.8 5.6 10.5 5.2 −10.9 −14.3 −12.4 −12.2 pCBA_ClO_C3_A 10.7 13.8 14.7 13.9 5.7 3.3 1.2 2.4 pCBA_Cl_C2_H 17.3 13.2 17.6 12.5 15.8 12.1 13.6 11.1 1) 反应路径为硝基苯、苯甲酸和对氯苯甲酸分别与3种自由基在不同位点的反应. 其中A代表加成反应,H代表抽氢反应.
1) Pathways mean that NB, BA and pCBA reacted with ·OH, ·ClO and ·Cl at different sites. A means radical addition pathway as well as H means hydrogen abstraction pathway. 下载: 导出CSV
下载: 导出CSV
表 2 3种化合物各位点与·OH的吉布斯自由能垒(ΔG0,≠,kcal·mol−1),吉布斯自由能变(ΔG0,kcal·mol−1)以及速率常数(ki,mol·L−1·s−1)
Table 2. Free energy barriers (ΔG0,≠, kcal·mol−1), reaction free energy (ΔG0, kcal·mol−1) and calculated rate constants (ki, mol·L−1·s−1) for NB+, BA and pCBA reaction with ·OH
反应位点
Sites·OH与NB+加成反应
·OH-addition of NB+ with ·OH·OH与NB+抽氢反应
H-abstraction of NB+ with ·OHΔG0,≠ ΔG0 ki ΔG0,≠ ΔG0 ki C1 16.9 −13.8 8.9 × 10 7.7 −6.2 1.3 × 109 C2 19.2 5.1 1.8 11.2 −18.1 3.4 × 106 C3 13.9 −10.8 1.4 × 104 11.5 −20.5 2.1 × 106 C4 20.1 −22.9 4.0 × 10−1 C5 13.5 −11.4 2.7 × 104 12.8 −19.7 2.3 × 105 C6 19.7 5.2 7.9 × 10−1 11.6 −20.9 1.8 × 106 ·OH与BA加成反应
·OH-addition of BA with ·OH·OH与BA抽氢反应
H-abstraction of BA with ·OHC1 7.9 −10.9 3.5× 108 6.4 −6.1 9.9 × 109 C2 9.2 −7.2 3.9× 107 8.0 −3.0 6.7 × 108 C3 10.1 −10.7 8.5× 106 5.6 −7.1 3.8 × 1010 C4 10.0 −4.5 1.0 × 107 C5 8.6 −11.3 1.1 × 108 5.9 −5.9 2.3 × 1010 C6 7.7 −8.8 4.9 × 108 7.4 −4.4 1.8 × 109 ·OH与pCBA加成反应
·OH-addition of pCBA with ·OH·OH与pCBA抽氢反应
H-abstraction of pCBA with ·OHC1 12.4 −11.8 1.8 × 105 C2 8.8 −8.2 7.6 × 107 5.6 −4.2 4.3 × 1010 C3 11.4 −9.4 9.5 × 105 5.9 −4.6 2.6 × 1010 C4 9.4 −5.0 2.8 × 107 C5 10.4 −10.6 5.4 × 106 5.2 −5.8 8.5 × 1010 C6 9.2 −7.9 3.9 × 107 5.5 −4.0 5.1 × 1010
下载: 导出CSV
表 3 3种化合物各位点与·ClO的吉布斯自由能垒(ΔG0,≠,kcal·mol−1),吉布斯自由能变(ΔG0,kcal·mol−1)以及速率常数(ki,mol·L−1·s−1)
Table 3. Free energy barriers (ΔG0,≠ , kcal·mol−1), reaction free energy (ΔG0, kcal·mol−1) and calculated rate constants (ki, mol·L−1·s−1) for NB+, BA and pCBA reaction with ·ClO
反应位点
Sites·ClO与NB+加成反应
Addition of NB+ with ·ClO·ClO与NB+抽氢反应
H-abstraction of NB+ with ·ClOΔG0,≠ ΔG0 ki ΔG0,≠ ΔG0 ki C1 15.4 −2.8 1.1× 103 22.1 13.5 2.7 × 10−2 C2 21.3 15.3 5.3 × 10−2 23.5 14.7 2.6 × 10−3 C3 12.4 −1.4 1.8 × 105 24.7 5.8 3.4 × 10−4 C4 25.1 −19.7 8.8 × 10−5 C5 12.2 −1.1 2.5 × 105 23.9 4.7 1.3 × 10−3 C6 20.9 17.2 1.0 × 10−1 24.5 15.1 4.7 × 10−4 ·ClO与BA加成反应
Addition of BA with ·ClO·ClO与BA抽氢反应
H-abstraction of BA with ·ClOC1 13.2 1.8 4.6 × 104 11.8 10.7 9.5 × 105 C2 13.7 4.6 2.0 × 104 13.4 10.2 6.4 × 104 C3 14.3 4.1 7.1 × 103 14.8 7.9 6.0 × 103 C4 15.5 8.7 9.4 × 102 C5 14.0 4.3 1.2 × 104 14.5 9.7 9.9 × 103 C6 13.0 3.8 6.4 × 104 14.2 13.7 1.6 × 104 ·ClO与pCBA加成反应
Addition of pCBA with ·ClO·ClO与pCBA抽氢反应
H-abstraction of pCBA with ·ClOC1 15.6 −0.7 7.9 × 102 C2 12.5 5.1 1.5 × 105 14.7 12.8 7.1 × 103 C3 13.7 3.8 2.0 × 104 15.8 9.5 1.1 × 103 C4 14.6 8.8 4.3 × 103 C5 13.3 3.6 3.8 × 104 14.2 9.4 1.6 × 104 C6 12.3 3.8 2.1 × 105 17.6 11.1 5.4 × 10
下载: 导出CSV
表 4 3种化合物各位点与·Cl的吉布斯自由能垒(ΔG0,≠,kcal·mol−1),吉布斯自由能变(ΔG0,kcal·mol−1)以及速率常数(ki,mol·L−1·s−1)
Table 4. Free energy barriers (ΔG0,≠ , kcal·mol−1), reaction free energy (ΔG0, kcal·mol−1) and calculated rate constants (ki, mol·L−1·s−1) for NB+, BA and pCBA reaction with ·Cl
反应位点
Sites·Cl与NB+抽氢反应
H-abstraction of NB+ with ·Cl·Cl与BA抽氢反应
H-abstraction of BA with ·Cl·Cl与pCBA抽氢反应
H-abstraction of pCBA with ·ClΔG0,≠ ΔG0 ki ΔG0,≠ ΔG0 ki ΔG0,≠ ΔG0 ki C1 14.1 7.4 9.2 × 103 5.4 3.2 2.1 × 1010 C2 10.5 6.0 4.0 × 106 10.4 9.5 4.7 × 106 10.2 7.3 6.6 × 106 C3 15.1 8.4 1.7 × 103 11.9 8.7 3.7 × 105 12.4 8.4 1.6 × 105 C5 15.7 8.1 6.2 × 102 10.3 9.4 5.6 × 106 10.9 8.9 2.0 × 106 C6 10.3 6.7 5.6 × 106 10.4 10.0 4.7 × 106 10.2 7.6 6.6 × 106
下载: 导出CSV
表 5 酸性条件下紫外/氯体系中3种化合物(NB+,BA和pCBA)分别与3种自由基的单电子转移反应能垒(ΔG0,≠,kcal·mol−1)及反应速率常数k(mol·L−1·s−1)
Table 5. Free energy barriers (ΔG0,≠ , kcal·mol−1) and the theoretical calculated rate constants (k, mol·L−1·s−1) for the electron transfer reaction between reactive radicals and three compounds (NB+, BA and pCBA )in UV/chlorine process at pH < 4.0
化合物
Compounds·OH ·ClO ·Cl ΔG0,≠ ki ΔG0,≠ ki ΔG0,≠ ki NB 10.8 1.9 × 106 17.6 1.9 × 10 11.3 8.1 × 105 BA 7.4 5.8 × 108 12.3 1.5 × 105 5.8 8.6 × 109 pCBA 8.3 1.3 × 108 13.1 3.9 × 104 8.2 1.5 × 108
下载: 导出CSV
表 6 酸性及中性条件下3种化合物分别与3种自由基的反应速率常数以及总反应速率常数kCP-OH,kCP-ClO和kCP-Cl
Table 6. The theoretical calculated rate constants (k, mol·L−1·s−1) for reactive radicals-initiated reactions of nitrobenzene (NB+), benzoic acid (BA) and p-chlorobenzoic acid (pCBA) in UV/chlorine process at pH < 4.0 and pH = 7.0
化合物
CompoundspH < 4.0 pH = 7.0 1) kCP-OH kCP-ClO kCP-Cl kCP-OH kCP-ClO kCP-Cl NB 1.3 × 109 4.3 × 105 1.0 × 107 1.7 × 1011 2.6 × 107 2.7 × 109 BA 9.0 × 1010 1.3 × 106 2.8 × 1010 1.1 × 1012 2.3 × 107 1.8 × 103 pCBA 1.3 × 1011 4.9 × 105 1.6 × 108 1.3 × 1012 3.1 × 107 3.2 × 108 1) 数据来源参考文献[32]. 1) Data obtained from reference[32].
下载: 导出CSV
-
[1] KRASNER S W. The formation and control of emerging disinfection by-products of health concern[J]. Philosophical Transactions. Series A, Mathematical, Physical, and Engineering Sciences, 2009, 367(1904): 4077-4095. [2] RICHARDSON S D, PLEWA M J, WAGNER E D, et al. Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: A review and roadmap for research[J]. Mutation Research/Reviews in Mutation Research, 2007, 636(1/2/3): 178-242. [3] YANG X, SUN J L, FU W J, et al. PPCP degradation by UV/chlorine treatment and its impact on DBP formation potential in real waters[J]. Water Research, 2016, 98: 309-318. doi: 10.1016/j.watres.2016.04.011 [4] 徐子文, 印红玲, 熊远明, 等. 紫外驱动高级氧化法降解水体中的磷酸三苯酯[J]. 环境科学, 2022, 43(11): 4992-4999. XU Z W, YIN H L, XIONG Y M, et al. Degradation of triphenyl phosphate in water by UV-driven advanced oxidation processes[J]. Environmental Science, 2022, 43(11): 4992-4999 (in Chinese).
[5] BULMAN D M, MEZYK S P, REMUCAL C K. The impact of pH and irradiation wavelength on the production of reactive oxidants during chlorine photolysis[J]. Environmental Science & Technology, 2019, 53(8): 4450-4459. [6] WATTS M J, LINDEN K G. Chlorine photolysis and subsequent OH radical production during UV treatment of chlorinated water[J]. Water Research, 2007, 41(13): 2871-2878. doi: 10.1016/j.watres.2007.03.032 [7] HUA Z C, GUO K H, KONG X J, et al. PPCP degradation and DBP formation in the solar/free chlorine system: Effects of pH and dissolved oxygen[J]. Water Research, 2019, 150: 77-85. doi: 10.1016/j.watres.2018.11.041 [8] WU Z H, FANG J Y, XIANG Y Y, et al. Roles of reactive chlorine species in trimethoprim degradation in the UV/chlorine process: Kinetics and transformation pathways[J]. Water Research, 2016, 104: 272-282. doi: 10.1016/j.watres.2016.08.011 [9] SUN P Z, LEE W N, ZHANG R C, et al. Degradation of DEET and caffeine under UV/chlorine and simulated sunlight/chlorine conditions[J]. Environmental Science & Technology, 2016, 50(24): 13265-13273. [10] XIANG Y Y, FANG J Y, SHANG C. Kinetics and pathways of ibuprofen degradation by the UV/chlorine advanced oxidation process[J]. Water Research, 2016, 90: 301-308. doi: 10.1016/j.watres.2015.11.069 [11] FORSYTH J E, ZHOU P R, MAO Q X, et al. Enhanced inactivation of Bacillus subtilis spores during solar photolysis of free available chlorine[J]. Environmental Science & Technology, 2013, 47(22): 12976-12984. [12] WANG W L, WU Q Y, HUANG N, et al. Synergistic effect between UV and chlorine (UV/chlorine) on the degradation of carbamazepine: Influence factors and radical species[J]. Water Research, 2016, 98: 190-198. doi: 10.1016/j.watres.2016.04.015 [13] CAI W W, PENG T, Yang B, et al. Kinetics and mechanism of reactive radical mediated fluconazole degradation by the UV/chlorine process: Experimental and theoretical studies[J]. Chemical Engineering Journal, 2020, 402: 126224. doi: 10.1016/j.cej.2020.126224 [14] LEI Y, YU Y F, LEI X, et al. Assessing the use of probes and quenchers for understanding the reactive species in advanced oxidation processes[J]. Environmental Science & Technology, 2023, 57(13): 5433-5444. [15] FANG J Y, FU Y, SHANG C. The roles of reactive species in micropollutant degradation in the UV/free chlorine system[J]. Environmental Science & Technology, 2014, 48(3): 1859-1868. [16] ZHOU S Q, ZHANG W Q, SUN J L, et al. Oxidation mechanisms of the UV/free chlorine process: Kinetic modeling and quantitative structure activity relationships[J]. Environmental Science & Technology, 2019, 53(8): 4335-4345. [17] CHUANG Y H, CHEN S, CHINN C J, et al. Comparing the UV/monochloramine and UV/free chlorine advanced oxidation processes (AOPs) to the UV/hydrogen peroxide AOP under scenarios relevant to potable reuse[J]. Environmental Science & Technology, 2017, 51(23): 13859-13868. [18] WANG D, BOLTON J R, HOFMANN R. Medium pressure UV combined with chlorine advanced oxidation for trichloroethylene destruction in a model water[J]. Water Research, 2012, 46(15): 4677-4686. doi: 10.1016/j.watres.2012.06.007 [19] MINAKATA D, KAMATH D, MAETZOLD S. Mechanistic insight into the reactivity of chlorine-derived radicals in the aqueous-phase UV-chlorine advanced oxidation process: Quantum mechanical calculations[J]. Environmental Science & Technology, 2017, 51(12): 6918-6926. [20] XIE H B, LI C, HE N, et al. Atmospheric chemical reactions of monoethanolamine initiated by OH radical: Mechanistic and kinetic study[J]. Environmental Science & Technology, 2014, 48(3): 1700-1706. [21] XIE H B, MA F F, WANG Y F, et al. Quantum chemical study on ·Cl-initiated atmospheric degradation of monoethanolamine[J]. Environmental Science & Technology, 2015, 49(22): 13246-13255. [22] ZHANG H Q, XIE H B, CHEN J W, et al. Prediction of hydrolysis pathways and kinetics for antibiotics under environmental pH conditions: A quantum chemical study on cephradine[J]. Environmental Science & Technology, 2015, 49(3): 1552-1558. [23] LI C, CHEN J W, XIE H B, et al. Effects of atmospheric water on ·OH-initiated oxidation of organophosphate flame retardants: A DFT investigation on TCPP[J]. Environmental Science & Technology, 2017, 51(9): 5043-5051. [24] LI F, BORTHWICK A G L, LIU W. Environmental theoretical calculation for non-periodic systems[J]. Trends in Chemistry, 2023, 5(6): 410-414. doi: 10.1016/j.trechm.2023.03.009 [25] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian, Inc[Z]. Wallingford CT, 2016. Gaussian 16, Revision B.01. [26] ZHAO Y, TRUHLAR D G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals[J]. Theoretical Chemistry Accounts, 2008, 120(1): 215-241. [27] SKYNER R E, McDONAGH J L, GROOM C R, et al. A review of methods for the calculation of solution free energies and the modelling of systems in solution[J]. Physical Chemistry Chemical Physics:PCCP, 2015, 17(9): 6174-6191. doi: 10.1039/C5CP00288E [28] OCHTERSKI J W. Thermochemistry in Gaussian[EB/OL].http://gaussian.com/thermo/ (Accessed 23 June 2020). [29] FU M C, SHANG R, ZHAO B, et al. Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide[J]. Science, 2019, 363(6434): 1429-1434. doi: 10.1126/science.aav3200 [30] ZHOU J, CHEN J W, LIANG C H, et al. Quantum chemical investigation on the mechanism and kinetics of PBDE photooxidation by ·OH: A case study for BDE-15[J]. Environmental Science & Technology, 2011, 45(11): 4839-4845. [31] XIA D M, CHEN J W, YU H, et al. Formation mechanisms of iodine-ammonia clusters in polluted coastal areas unveiled by thermodynamics and kinetic simulations[J]. Environmental Science & Technology, 2020, 54(15): 9235-9242. [32] PENG T, XU C, YANG L, et al. Kinetics and mechanism of degradation of reactive radical-mediated probe compounds by the UV/chlorine process: Theoretical calculation and experimental verification[J]. ACS Omega, 2022, 7(6): 5053-5063. doi: 10.1021/acsomega.1c06001 [33] AN T C, AN J B, GAO Y P, et al. Photocatalytic degradation and mineralization mechanism and toxicity assessment of antivirus drug acyclovir: Experimental and theoretical studies[J]. Applied Catalysis B:Environmental, 2015, 164: 279-287. doi: 10.1016/j.apcatb.2014.09.009 [34] ZHANG Y N, WANG J Q, CHEN J W, et al. Phototransformation of 2, 3-dibromopropyl-2, 4, 6-tribromophenyl ether (DPTE) in natural waters: Important roles of dissolved organic matter and chloride ion[J]. Environmental Science & Technology, 2018, 52(18): 10490-10499. [35] GUO K H, WU Z H, CHEN C Y, et al. UV/chlorine process: An efficient advanced oxidation process with multiple radicals and functions in water treatment[J]. Accounts of Chemical Research, 2022, 55(3): 286-297. doi: 10.1021/acs.accounts.1c00269 [36] CAI W W, PENG T, ZHANG J N, et al. Degradation of climbazole by UV/chlorine process: Kinetics, transformation pathway and toxicity evaluation[J]. Chemosphere, 2019, 219: 243-249. doi: 10.1016/j.chemosphere.2018.12.023 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 1397
- HTML全文浏览数: 1397
- PDF下载数: 16
- 施引文献: 0









