


-
加氢催化剂是石油冶炼过程中最重要的催化剂之一,主要用于重油的脱硫脱氮等[1]。加氢催化剂在使用过程中,由于高温以及有害物质的沉积吸附等影响,会永久性失活[2]。因此,不可避免地要置换出大量的废催化剂[3]。废加氢催化剂主要是由贵金属钒、钼、镍、钴等和载体氧化铝组成。因催化反应的需要,催化剂在制作过程中不得不添加一些有毒的组分,如As2O3、As2O5、CrO3等,这些有毒物质会沉积在废催化剂中[4]。倘若对废催化剂不加处置随意堆放的话,一方面堆积废催化剂需要占据大量的场地;另一方面废催化剂中的有毒物质会对环境造成极大的危害[4]。而废加氢催化剂中的贵金属钒 (V) 、钼 (Mo) 、镍 (Ni) 都是重要的战略金属,可被广泛应用于国民经济和国防军工等领域[5],故具有重要的回收价值。所以,实现废加氢催化剂的资源化利用,对环境保护和石油工业的可持续发展都是十分必要的[6]。
目前,Mo-Ni系催化剂是使用最多的一类加氢催化剂,针对这一类催化剂的有价金属回收方法主要有焙烧-浸出法、浸出-萃取法、浸出-离子交换法、加压碱浸法、氨浸法、生物浸出法等方法[7]。这些方法的共同点都是先将有价金属Mo、Ni、铝 (Al) 等从固相中转移到液相中,再在液相中对有价金属进行富集回收。因此,如何高效地将有价金属从固相中转移到液相中是关键技术问题。Mo、Al和Ni的化学性质不同,可选用钠化焙烧-水浸的方法将Mo、Al转化为可溶态,与Ni浸出分离。但是,目前并没有关于钠化焙烧-浸出过程的详细研究。
本研究拟采用响应面曲线法考察钠化焙烧-浸出过程中的碳酸钠添加量、焙烧温度、时间和浸出液固比、浸出温度对Mo、Al浸出率的影响;并且,结合热重-差示扫描量热法(TG-DSC)和X射线衍射法分析钠化焙烧过程中Mo、Al的转化机理。
-
供试原料是从某石油化工厂回收的废加氢催化剂,为黑色条状颗粒。经球磨化学法处理后采用X射线荧光光谱仪 (XRF) 对其进行分析,结果见表1。由表1可知,催化剂中载体氧化铝的质量分数为47.82%,含量最高;MoO3和NiO的质量分数分别为21.71%和3.558%,钼、镍含量亦较高,有很大的回收价值。
-
1) 焙烧预处理。废催化剂在使用过程中,表面有大量的积碳和油脂,所以需对废催化剂进行焙烧预处理,并使废催化剂中的金属化学形态从硫化物转化为氧化物[8]。
2) 钠化焙烧-水浸法。取10 g经焙烧预处理后的废加氢催化剂,按比例添加一定量的碳酸钠,充分研磨,使其混合均匀。将混匀后的样品置于马弗炉内,在一定温度下进行钠化焙烧[7]。主要反应式如式 (1)~式 (3) 所示。
样品经钠化焙烧后,加入一定量的水溶于锥形瓶中,置于磁力加热搅拌器上,恒温浸出。反应结束后,待样品冷却,将反应物进行抽滤,滤液存于离心管中。浸出液中钼含量测定采用硫氰酸盐比色法[9],铝含量测定采用铝试剂法。根据浸出液中Mo和Al的质量浓度,计算出Mo和Al的浸出率;焙烧过程的机理分析利用热重-差示扫描仪和X射线衍射仪进行分析。
3) 响应面法实验设计。响应曲面法设计软件为Design-expert 8.0.6;选择钠化焙烧温度(A)、焙烧时间(B)、碳酸钠添加量与催化剂质量比(简称为“碳酸钠添加量”;C)、浸出液固比(D)和浸出温度(E)为5个因素,以Mo和Al的浸出率为响应值,运用中心组合实验设计 (BBD) 法进行5因素3水平的响应面分析实验。设计的实验因素水平及编码见表2。按表2列出的实验因素水平,利用Design Expert软件,采用Box-Behnken模型进行实验设计,对实验结果进行多元回归拟合,可得响应曲面编码形式的二次多项回归方程。
-
对废催化剂进行焙烧预处理的目的是去除其表面的积碳和油脂。取10 g废催化剂分别在不同的温度下焙烧2 h,考察废催化剂烧失量的变化,结果见表3。由表3可知,随着焙烧温度增加,废催化剂的烧失量逐渐增加。当预处理温度较低时,废催化剂表面的油脂去除不完全,烧失量较低。因MoO3的熔点为795 ℃[10],故当预处理温度过高时,可使部分MoO3发生升华,所以预处理温度不宜高于800 ℃。不同温度预处理后的废催化剂进行X射线衍射分析的结果如图1所示。当预处理温度为300 ℃时,废催化剂表面的油脂和碳仍有大量残留,会影响X射线衍射分析,此时样品特征峰不明显,杂峰较多。当预处理温度在400~500 ℃时,预处理后的废催化剂主要物相为MoO3、Al2O3;MoO3、Al2O3均为两性氧化物,他们能与碱及某些强酸反应,从而方便后续的钠化焙烧实验[11];当预处理温度为600~800 ℃时,X射线衍射图中已无MoO3峰,废催化剂的主要物相为Al2(MoO4)3,这说明MoO3和Al2O3在高温下会结合生成Al2(MoO4)3。
为了探究Al2(MoO4)3参与钠化反应的过程及机理,分别取在500、600、700、800 ℃预处理后的废催化剂分别添加其质量1.1倍的碳酸钠充分磨匀混合,在950 ℃下焙烧2 h,待物料冷却后进行X射线衍射分析,结果如图2所示。由图2可知,经过不同温度预处理后的废催化剂与碳酸钠钠化焙烧反应产物均为Na2MoO4和NaAlO2,这说明以理温度在600~800 ℃预处理时,MoO3、Al2O3结合生成的Al2(MoO4)3不会影响Mo、Al的钠化反应。但考虑到实际生产过程的能耗问题,废催化剂的适合预处理条件确定为400~500 ℃下焙烧2 h。
-
对响应面法得到的回归方程进行了方差分析和显著性检验,结果见表4。其中,P值的大小表明模型及各考察因素的显著水平,P值小于0.05,表明模型或该因素有显著影响;P值小于0.001,表明模型或各因素影响显著。失拟项用来表示所用模型与实验拟合的程度,即二者差异的程度[12]。由表4可知,FMo=15.97,FAl=10.00,P值均小于0.0001,说明该模型的拟合效果良好。失拟项PMo=0.3038>0.05,PAl=0.1427>0.05,说明二次多项式拟合方程与实际数据吻合。
对于响应值E(Mo),参数A、B、C、E、A2、B2、C2、D2、E2的效应显著 (P<0.05) ;模型决定系数R2=0.9274,变异系数CVqe=1.48%<4%,表明模型的可信度和精确度较高[13];精密度是有效信号与噪声的比值,大于4即视为合理;对于E(Mo)的分析,本实验的信噪比为14.459,表示信号充足。对于响应值E(Al),参数A、B、C、A2、B2、C2、D2的效应显著 (P<0.05) ;模型决定系数R2=0.8889,变异系数CVqe=2.12%<4%,表明模型的可信度和精确度较高;对于E(Al)的分析,本实验的信噪比为11.680,表示信号充足。综合以上信息,可判断此模型拟合效果较好,可用于预测。
-
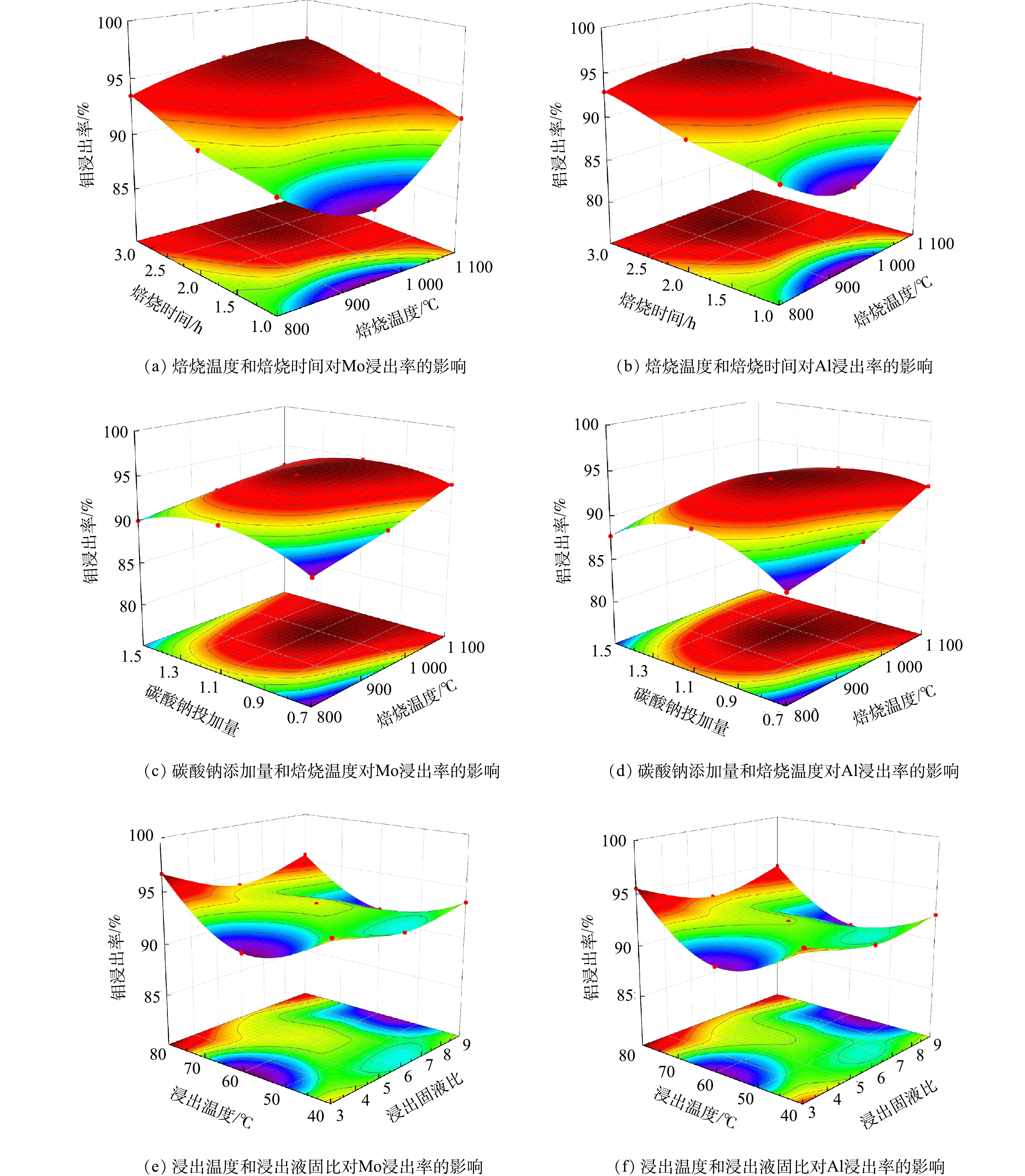
对模型中各项参数进行显著性检测发现,焙烧温度、焙烧时间、碳酸钠添加量对Mo、Al浸出率有显著影响 (P<0.05) 。在交互项中,各因素之间的交互作用均不明显[14]。由图3 (a) 、3 (b) 可以看出,Mo、Al浸出率随着焙烧温度和焙烧时间的提高均显著增加。当焙烧温度为800 ℃、焙烧时间也较低时,钠化反应不充分,Mo、Al的浸出率均在80%左右,且Al的浸出率略低于Mo的浸出率。随着焙烧温度的提高,Mo、Al浸出率均显著增加。焙烧温度也不宜过高,超过1 000 ℃下焙烧,物料易挥发,Mo、Al的浸出率反而有所下降。
碳酸钠添加量对Mo、Al浸出率的影响均较为显著。由于废催化剂中的硅、磷、砷等杂质也会消耗部分碳酸钠,所以加入的碳酸钠要过量。由图3 (c) 、3 (d) 可以看出,当碳酸钠添加量与废催化剂质量比大于1时,Mo、Al才能充分浸出;但碳酸钠的添加量再增加时,碳酸钠与P、Si等反应生成杂质就会增多,导致物料易烧结,从而会影响Mo、Al的浸出。
由图3 (e) 、3 (f) 可知,Mo、Al浸出率随着浸出温度升高而升高。提升温度可以增强溶液中离子活性,增大反应速率,当其他因素一定时,浸出温度在70 ℃时就能完成浸出过程;随着浸出液固比的增大,Mo、Al浸出率都呈先上升后下降的趋势。当浸出液固比为3∶1时,烧料与液体接触面积小,浸出反应不充分;但当浸出液固比过大时,则溶液碱性降低,也不利于Mo、Al浸出。所以,浸出液固比在6∶1左右就能确保Mo、Al的充分浸出。
-
根据回归模型,预测出最佳工艺条件为:焙烧温度为954 ℃、焙烧时间为2 h、碳酸钠添加量(Na2CO3与废加氢催化剂质量比)为1.1、浸出液固比为6∶1、浸出温度为70 ℃。在该条件下,Mo的浸出率在98.79%,Al浸出率为94.54%。
在最佳工艺条件下进行3组平行验证实验,结果见表5。Mo浸出率在98%以上、Al浸出在94%以上。浸出液中,Al质量浓度在30 g·L−1左右、Mo质量浓度为18 g·L−1左右,这说明响应面优化结果对实际生产具有一定的参考价值。
-
1) 碳酸钠添加量对Mo、Al钠化反应的影响。图4为经预处理后的催化剂添加不同量碳酸钠焙烧产物 (950 ℃) 的物相分析。由图4可知,在不同碳酸钠添加量下,焙烧产物的主要成分是钼酸钠和偏铝酸钠;碳酸钠添加量与废催化剂量比为0.7~0.9时,XRD图谱中有Na2MoO4、NaAlO2、Al2O3、Al2(MoO4)3衍射峰,此时碳酸钠量不足,部分MoO3与Al2O3结合生成了Al2(MoO4)3;当碳酸钠添加量与废催化剂量比为1.0时,焙烧产物中废催化剂中Al2(MoO4)3峰消失,但仍有Al2O3峰,这说明此时碳酸钠添加量仍不足;碳酸钠添加量与废催化剂量比为1.1、1.2时Al2O3峰也消失,这说明此时碳酸钠添加量已足够,几乎全部的铝也与钠盐反应。
图5为当固定浸出液固比为6:1、浸出温度为60 ℃时,Mo、Al浸出率与碳酸钠添加量的变化关系。由图5可知,Mo、Al浸出率总体呈先上升后下降的趋势;在碳酸钠添加量小于1.1倍时,Mo、Al浸出率随着碳酸钠添加量的增加而增加;当碳酸钠添加量为1.1倍时,Mo、Al浸出率最高,分别为98.75%、95.02%;但当碳酸钠添加量大于1.1倍时,Mo、Al浸出率又有所降低。这是因为,随着碳酸钠的过量加入会影响物料的熔点,物料易烧结,导致Mo、Al浸出率降低。综合上述信息可知,碳酸钠加入量为废催化剂质量的1.1倍较合适。
2) 焙烧温度对Mo、Al钠化反应的影响。图6是在500 ℃ (空气气氛,升温速率10 ℃·min−1) 下,焙烧预处理后的废加氢催化剂中加入其质量比为1.1倍的碳酸钠后的TG-DSC曲线。在100、750 ℃处出现放热峰,这说明有明显的的质量损耗。第1个放热峰是因为样品中水分的蒸发;第2个峰在700~1000 ℃之间,此时因MoO3、Al2O3参与钠化反应,生成CO2产生质量减少的现象。由此可知,钠化反应大量发生的温度为700~100 0 ℃。由图7可知,催化剂在不同焙烧温度下的钠化焙烧产物物相均为Na2MoO4、NaAlO2。在700 ℃时,Na2MoO4、NaAlO2峰较弱,此时钠化反应未大量发生;800~950 ℃时,Na2MoO4、NaAlO2峰显著增强;在950 ℃时Na2MoO4、NaAlO2峰达到最强,这说明在此温度下钠化反应大量发生。随着焙烧温度的进一步升高,Na2MoO4、NaAlO2峰又有所降低,这是因为过高温度下物料易挥发,使得产物减少。
图8为在固定浸出液固比为6:1、浸出温度为60 ℃条件下,Mo、Al浸出率与钠化焙烧温度的变化关系。由图8可知,当温度较低时,钠化反应未大量发生,Mo、Al浸出率较低;当温度达到950 ℃时,Mo、Al浸出率最高,分别为98.11%、94.87%。超过1 000 ℃时,Mo、Al浸出率又有所下降。这是因为,过高温度钼会挥发损失,物料也会发生烧结,影响Mo、Al的浸出。综合上述信息可知,钠化反应最佳温度为950 ℃。
3)焙烧时间对Mo、Al钠化反应的影响。由图9可知,不同焙烧时间下烧料的物相均为Na2MoO4和NaAlO2。当焙烧时间为1、1.5 h时,由于反应时间较短,所以Na2MoO4和NaAlO2峰较弱;当焙烧时间为2 h时,Na2MoO4和NaAlO2峰显著增强;继续增加焙烧时长,Na2MoO4和NaAlO2峰则并无明显变化。
图10为在固定浸出液固比为6:1、浸出温度为60 ℃条件下,Mo、Al浸出率与焙烧时间的变化关系。由图10可知,当焙烧时间小于2 h时,Mo、Al浸出率随着焙烧时间的增加而增加;焙烧时间为2 h时,Mo、Al浸出率最大,分别为98.22%、94.03%;当焙烧时间大于2 h时,铝的浸出率变化不大,钼浸出率明显下降。这是因为,随着焙烧时间的增加,部分钼挥发损失,物料也易发生烧结,从而不利于金属的浸出。综合上述信息可知,钠化焙烧时间为2 h较合适。
-
1) 适宜的废催化剂焙烧预处理温度为400~500 ℃。随着预处理温度的升高,钼与铝会结合生成Al2(MoO4)3;Al2(MoO4)3也可以参与钠化反应。
2) 基于响应面得出的钠化焙烧-水浸回收废加氢催化剂中有价金属的最优工艺条件为:焙烧温度950 ℃、焙烧时间2 h、Na2CO3与废加氢催化剂质量比1.1∶1、浸出液固比6∶1、浸出温度70 ℃;在此条件下,Mo浸出率超过98%、浸出率超过94%。
3) 碳酸钠添加量较少时,未参与钠化反应的钼和铝会结合生成Al2(MoO4)3;而碳酸钠添加过量时,物料易烧结,造成Mo、Al浸出率降低。
4) Mo、Al发生钠化反应的温度在700~1 000 ℃之间;继续提高温度会加大物料挥发,导致生成的Na2MoO4、NaAlO2减少,Mo、Al浸出率降低。
基于响应曲面法的钠化焙烧-水浸法回收废加氢催化剂中有价金属工艺优化
Optimization of recovery of valuable metals from spent hydrogenation catalyst by sodium roasting water leaching method based on response surface methodology
-
摘要: 为提高废加氢催化剂钠化焙烧-水浸分离工艺回收有价金属的效率,采用响应面法优化了废加氢催化剂钠化焙烧-水浸工艺条件,探讨了焙烧温度、焙烧时间、碳酸钠添加量、浸出温度和液固比对Mo、Al浸出率的影响,并确定了最优工艺条件;同时,采用热重-差示扫描量热法 (TG-DSC) 和X射线衍射法 (XRD) 分析了废催化剂预处理与钠化焙烧过程的物相变化。结果表明:适宜的预处理温度为400~500 ℃。随着预处理温度的升高,钼与铝结合生成Al2(MoO4)3;Al2(MoO4)3可参与钠化反应。在钠化焙烧温度为950 ℃、焙烧时间为2 h、碳酸钠与废加氢催化剂质量比为1.1∶1、浸出液固比为6:1、浸出温度为70 ℃的条件下,Mo的浸出率可达98%以上、Al的浸出率可达94%以上。当碳酸钠添加量较少时,未参与钠化反应的Mo和Al会结合生成Al2(MoO4)3,从而造成Mo、Al浸出率降低。Mo、Al发生钠化反应的温度在700~1000 ℃之间;当焙烧温度为700 ℃时,钠化反应未大量发生;随着反应温度的提高,钠化反应大量发生,至950 ℃时钠化反应最充分。本研究结果可为废加氢催化剂钠化焙烧-水浸分离回收有价金属提供参考。Abstract: The process conditions of sodium roasting and water extraction were optimized by response surface method during the recovery of valuable metals from spent hydrogenation catalyst. The effects of roasting temperature, roasting time, sodium carbonate adding amount, leaching temperature and liquid-solid ratio on the extraction rate of Mo and Al were examined, and the optimal process conditions were determined. TG-DSC and X-ray diffraction were used to analyze the phase transformation during pretreatment and sodium roasting of spent catalyst. The results showed that the suitable pretreatment temperature was located in the range of 400 and 500 ℃. With the increase of pretreatment temperature, Al2(MoO4)3 can be formed by the combination of Mo and Al, while Al2(MoO4)3 can still participate in the sodium reaction. The maxmium extraction ratios of Mo and Al of over 98% and 94% could be obtained when the mixture of sodium carbonate and spent hydrogenation catalyst with mass ratio of 1.1:1 was roasted at 950 ℃ for 2 h, and was further extracted by water with the liquid-solid ratio of 6:1 at 70 ℃. Mo and Al combine and form Al2(MoO4)3 with the introduction of slight sodium carbonate. The sodium reaction did not occur substantially at 700 ℃, while finish completely with the temperature rising to 950 ℃. This study provides technical and theoretical supporting for the sodium roasting and water extraction process of spent hydrogenation catalyst.
-
作为一种具有高毒性、生物累积和生物放大效应的重金属,汞(Hg)主要以气态形式被排放到大气中,随后可通过大气环流进行全球传输并沉降到陆地和海洋生态系统. 化石燃料燃烧、金属冶炼和垃圾焚烧等人为活动每年产生2000—3000 t的汞排放,而地质活动等自然源的每年汞排放量可达5500 t[1]. 目前,在无明显人为源的偏远地区也普遍检出汞[2],如青藏高原的海螺沟冰川融水中汞含量为6.96—10.78 ng·L-1[3],北极苔原也有显著大气汞沉降[4]. 为降低汞污染对生态系统与人体健康的危害,2013年世界上128个国家和地区签署了《关于汞的水俣公约》以减少汞的使用与人为排放[5],2018年全球人为导致的汞排放较2013年已出现下降[6]. 然而,由于历史上累积汞在表生环境中的再释放和循环,汞污染问题仍将长期存在[7]. 此外,全球变暖可能导致冰川与冻土中汞的释放[8],而升温引起的生态系统中初级生产力提高、生物习性改变以及食物网结构的变化也可能加剧生物体中汞的累积[9].
引起水俣病等公害事件的甲基汞(MeHg)是毒性最高的汞形态之一,其主要来源于硫酸盐还原菌、铁还原菌和产甲烷菌等含有hgcAB基因簇微生物的甲基化[10]. 甲基汞通过与含巯基的蛋白质结合,对生物体造成以神经系统为主的全身性损伤,并可跨过胎盘屏障导致先天性疾病[11-13]. 甲基汞的健康风险与甲基汞暴露直接相关,如食用鱼肉等水产品是甲基汞的重要暴露途径[12,14]. 尽管水中甲基汞在总汞中的占比通常较低,但鱼体中甲基汞可达总汞的95%左右[15],该现象主要是因为甲基汞的生物累积和生物放大效应. 作为海洋中的初级生产者,藻类具有极强的汞富集能力,部分藻中汞浓度可达水环境的103—106倍[16]. 藻类富集的甲基汞可通过食物链传递并在其他生物体内累积,甲基汞的营养级放大斜率可达0.15 —0.35,表现出显著的生物放大效应[17-20]. 同时,日趋严重的全球变暖、水体酸化和富营养化等问题带来了环境因子、藻类丰度、汞浓度与可利用性的新变化.
开展甲基汞在藻类中的富集过程研究以及这一过程对水体甲基汞生物累积与放大的关键影响研究对于揭示汞的生物富集、传递特性以及预测其风险至关重要. 因此,本文对藻类富集甲基汞的特征、机理及影响因素进行详细讨论,总结甲基汞在藻类中的分布、转化与后续营养传递特征,并对相关研究的发展方向进行展望.
1. 藻类富集甲基汞的特征、机制和影响因素(The patterns, mechanisms, and influencing factors of methylmercury bioconcentration in algae)
藻类对甲基汞的富集涉及吸收、吸附、转运、解吸和外排等多个过程(图1),这些过程对甲基汞在藻类中的累积和食物链传递有重要影响. 首先,藻类可通过吸附、吸收、解吸和外排影响进入食物链的甲基汞的量;此外,藻类及其伴生菌与胞外分泌物可能通过去甲基化或甲基化改变食物链中甲基汞浓度;再者,甲基汞在藻体的不同亚细胞结构中的分布会影响甲基汞在食物链中的传递行为.
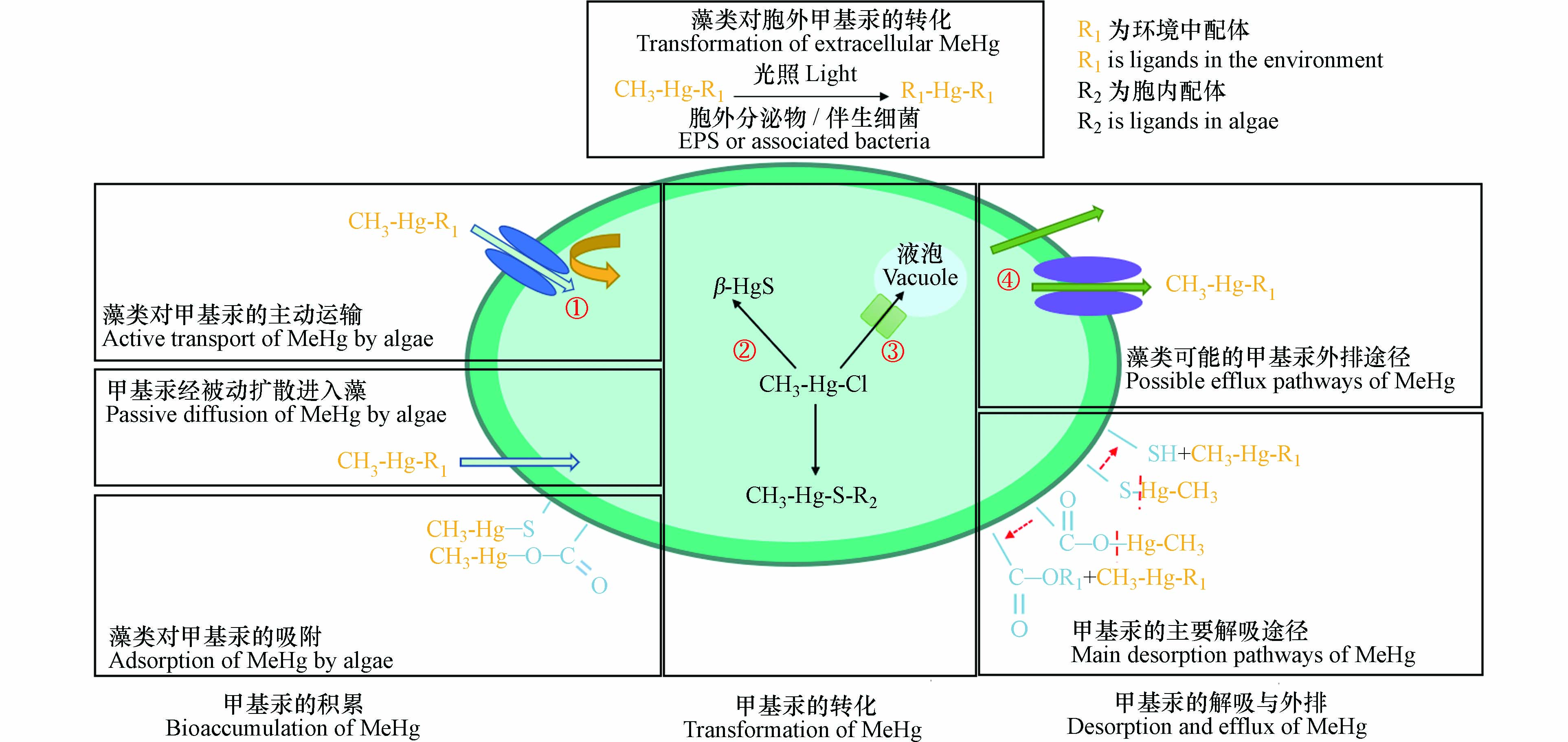 图 1 藻类吸附、吸收、转化、解吸与外排甲基汞的相关机制Figure 1. The proposed mechanisms of adsorption, absorption, transformation, desorption and efflux of MeHg by algae(① 未确定甲基汞主动运输的转运蛋白;② 未确定甲基汞是否可以转化为硫化汞颗粒;③ 未确定甲基汞进入液泡的途径;④ 未确定甲基汞的解吸与外排)(① The transporter of MeHg via active transport has not been identified; ② It is unknown whether MeHg can be converted to β-HgS; ③ The pathway of MeHg entering the vacuole has not been determined; ④ The desorption and efflux of MeHg are not determined)
图 1 藻类吸附、吸收、转化、解吸与外排甲基汞的相关机制Figure 1. The proposed mechanisms of adsorption, absorption, transformation, desorption and efflux of MeHg by algae(① 未确定甲基汞主动运输的转运蛋白;② 未确定甲基汞是否可以转化为硫化汞颗粒;③ 未确定甲基汞进入液泡的途径;④ 未确定甲基汞的解吸与外排)(① The transporter of MeHg via active transport has not been identified; ② It is unknown whether MeHg can be converted to β-HgS; ③ The pathway of MeHg entering the vacuole has not been determined; ④ The desorption and efflux of MeHg are not determined)1.1 甲基汞在藻类中的累积规律与机制
藻类可显著累积水体甲基汞,基于野外数据建立的模型显示浮游植物对甲基汞的生物累积因子(bioaccumulation factor,BAF)达102.4—105.9[21]. 由于实际环境中浮游生物是由多种藻类组成,难以确定不同藻类对甲基汞富集的具体贡献,所以藻类对甲基汞的富集研究通常选择环境中的硅藻与绿藻等优势藻种在实验室条件下开展工作[22]. 该部分论述了藻对甲基汞的累积规律,以及相关的吸附与吸收机制.
1.1.1 甲基汞在藻类中的累积规律
藻类累积甲基汞研究的主要指标包括藻类的体积富集因子(volume concentration factor,VCF)、富集率以及累积速率(具体计算方法见表1注). 表1比较了不同研究结果,其中,甲基汞的形态和暴露时间是影响藻类富集甲基汞的重要因素. 首先,不同形态甲基汞在藻类中的富集存在差异,相较于氯化甲基汞,半胱氨酸与蛋氨酸与甲基汞的络合会减弱藻类对甲基汞的富集[23-24]. 此外,藻类对甲基汞的富集具有一定的时间变化规律,通常在初始5 min内非常迅速,在24 h左右达到平衡[25],因此暴露24 h可测定平衡状态下藻类的富集率等指标,暴露1 h内可反映短期的甲基汞富集动力学变化.
表 1 实验室条件下常见藻类对甲基汞的富集效果Table 1. Bioconcentration of MeHg in common algae under laboratory conditions所属纲Class 受试藻类Subject algae 甲基汞形态MeHg species 甲基汞浓度/(nmol·L−1)Concentration 暴露时间/hExposure time 是否清洗Whether cleaned 吸收速率/(cell−1 h−1·nmol·L−1)aAbsorption rate lgVCFb/生物累积因子lgVCF/BCF 富集率cEnrichment ratio 文献References 绿藻纲 葡萄鼓藻Cosmarium botrytis CH3HgCl 0.02 24e 否 —f 5.94 ± 0.69b —f [30] 绿藻纲 钙化裂须藻Schizothrix calcicola CH3HgCl 0.02 24d 否 —f 5.60 ± 0.21b —f [30] 绿藻纲 小球藻Chlorella autotrophica CH3HgCl 3.29 72g 8 mmol·L−1半胱氨酸pH = 8.2,1 min —f 113.8h —f [31] 绿藻纲 羊角月牙藻Selenastrum capricornutum CH3HgCl 1.5 1 否 1.3×10−4 —f 0.97 [23] 绿藻纲 羊角月牙藻Selenastrum capricornutum CH3Hg-GSH 1.5i 1 否 1.03×10−4 —f 0.93 [23] 绿藻纲 羊角月牙藻Selenastrum capricornutum CH3HgCl 0.02 24e 否 —f 6.67 ± 0.13b —f [30] 绿藻纲 羊角月牙藻Selenastrum capricornutum CH3Hg-Cys 1.5i 1 否 1.11×10−4 —f 0.92 [23] 绿藻纲 蛋白核小球藻Chlorella pyrenoidsa CH3HgCl 0.4 24e 0.8 mmol·L−1半胱氨酸pH = 7.1,5 min 0.08 —f 0.46 [28] 绿藻纲 杜氏藻Dunaliella tertiolecta CH3HgCl 0.29—0.42 4d 否 0.63 ± 0.03 4.46b —f [32] 绿藻纲 斜生栅藻Scenedesmus obliquus CH3HgCl 0.4 24e 0.8 mmol·L−1半胱氨酸pH = 7.1,5 min 0.02 —f 0.86 [28] 蓝藻纲 聚球藻Synechococcus bacillaris CH3HgCl 0.29—0.42 4d 否 0.97±0.03 6.34b —f [32] 硅藻纲 海链藻Thalassiosira sp. CH3HgCl 0.02 24e 否 —f 5.37 ± 0.04b —f [30] 硅藻纲 假微型海链藻Thalassiosira pseudonana CH3Hg-Cys 4×10−3 1d 否 11—14×10−3 5.47b —f [32] 硅藻纲 假微型海链藻Thalassiosira pseudonana CH3HgCl 0.29—0.42 4d 否 15.00 ± 3.00 5.44b —f [32] 硅藻纲 假微型海链藻Thalassiosira pseudonana CH3Hg-Met 0.28—0.48 l 4 否 16.9 ± 0.2 6.22b —f [24] 硅藻纲 假微型海链藻Thalassiosira pseudonana CH3Hg-HA 0.28—0.48l 4 否 11.9 ± 1.5 6.07b —f [24] 硅藻纲 假微型海链藻Thalassiosira pseudonana CH3HgCl 7.32 72g 8 mmol·L−1半胱氨酸pH = 8.2,1 min —f 14.7h —f [31] 硅藻纲 威氏海链藻Thalassiosira weissflogii CH3HgCl 1.85 1d 8 mmol·L−1半胱氨酸pH = 8.2,1 min 4.6 9.8×106 j —f [33] 硅藻纲 威氏海链藻Thalassiosira weissflogii CH3HgCl 1.85 1k 8 mmol·L−1半胱氨酸pH = 8.2,1 min 3.01 7.4×106 j —f [33] 硅藻纲 布氏双尾藻Ditylum brightwellii CH3HgCl 0.1 1d 否 130 5.49b —f [34] 硅藻纲 旋链角毛藻Chaetoceros curvisetus CH3HgCl 2.5 5d 否 1.8 6.04b —f [34] 鞭藻纲 等鞭金藻Isochrysis galbana CH3HgCl 4.59 72g 8 mmol·L−1 半胱氨酸pH = 8.2,1 min —f 79.23h —f [31] 注:GSH: Glutathione; Cys: Cysteine; Met: Methionine; HA: Humic acid. (a. 单位为cell−1 h−1 nmol·L−1;b.体积富集因子(Volume concentration factor,VCF)=平衡后藻类中的甲基汞浓度(mol·µm−3)/水中的甲基汞浓度(mol·µm−3);c.富集率 = 平衡后藻类中的甲基汞含量(µg)/水中的甲基汞含量(µg);d.培养过程为全程光照条件;e.培养中明暗时间比为12 : 12;f.文章内未给出具体数据,用“—”表示;g.培养中明暗时间比为14 : 10;h.生物富集因子(Bioaccumulation factor,BAF)= 平衡后藻类中的甲基汞浓度(µg·g−1 湿重)/水中的甲基汞浓度(µg·g−1);i.藻类暴露甲基汞浓度为1.5 nmol·L−1,表中谷胱甘肽或半胱氨酸的浓度为90 nmol·L−1;j.生物富集因子 = 平衡后藻类中的甲基汞浓度(µg·mol−1 以生物炭计)/水中的甲基汞浓度(µg·L−1);k.藻在暴露前在2 µg·L−1的甲基汞溶液中驯化18 d(明暗时间比为14 : 10),大约为第18代藻类;l.暴露的蛋氨酸浓度为1 nmol·L−1,胡敏酸浓度为1 mg·L−1 C) (a. cell−1 h−1 nmol−1; b. VCF = MeHg concentration in algae (mol·µm−3) / MeHg concentration in water (mol·µm−3) after equilibrium; c. The enrichment rate = MeHg concentration in algae (µg) / MeHg concentration in water (µg) after equilibrium; d. The cultivation process was under full light; e. The ratio of light and dark time in culture was 12 : 12; f. No specific data is given in the reference, which is indicated by "-"; g. The ratio of light and dark time in culture was 14 : 10; h. BAF = MeHg concentration in algae (µg·g−1 wet weight) / MeHg concentration in water (µg·g−1) after equilibrium; i. The concentration of MeHg exposed to algae is 1.5 nmol·L−1, and the concentration of glutathione or cysteine in the table is 90 nmol·L−1; j. Bioconcentration factor = MeHg concentration in algae (µg·mol−1 in biochar) / MeHg concentration in water (µg·L−1) after equilibrium; k. Algae were acclimated in 2 µg·L−1 MeHg solution for 18 days before exposure (the ratio of light and dark time was 14:10), which was about the 18th generation of algae; l. The exposed methionine concentration was 1 nmol·L−1 and the humic acid concentration was 1 mg·L−1 C.) | Show Table DownLoad:
CSV
DownLoad:
CSV
藻类富集甲基汞主要通过吸收和吸附两种途径[26]. 一些研究采用乙二胺四乙酸(EDTA)或半胱氨酸等络合剂清洗藻类表面吸附的甲基汞,用以区分藻类通过吸附与吸收富集的甲基汞. 其中,0.8 mmol·L−1半胱氨酸清洗5 min或8 mmol·L−1半胱氨酸清洗1 min,藻类吸附甲基汞的解吸率可达90%以上[27]. 研究发现,藻类富集甲基汞的差异可能来源于藻类对甲基汞吸附与吸收的比例不同. 如5 ng·L−1 的甲基汞暴露下,斜生栅藻(Scenedesmus obliquus)平衡后的吸附率(藻吸收的甲基汞量/水体甲基汞量)为16.3%,低于蛋白核小球藻(Chlorella pyrenoidosa)(28.2%),但二者的吸附率(藻吸附的甲基汞量/水体甲基汞量)接近,且随甲基汞暴露浓度增加,斜生栅藻吸收率的增加幅度显著高于蛋白核小球藻[28]. 另外,暴露时间由24 h增加至168 h,蛋白核小球藻的吸附率由21.2%下降至11.3%,吸收率则由38.3%上升至46.9%[28]. 由于吸附速率大于吸收速率,推测藻类富集甲基汞前期以胞外快速吸附为主,平衡后通过胞内缓慢吸收进行富集[29].
1.1.2 藻类对甲基汞的吸附机制
藻类累积甲基汞的反应动力学研究发现水华束丝藻(Aphanizomenon flosaquae)与铜绿微囊藻(Microcystis aeruginosa)对甲基汞的吸附均较好地符合准二级动力学模型(R2 > 0.99),提示这一过程可能涉及物理吸附与化学吸附,化学吸附可能涉及藻类表面基团与甲基汞间的电子转移或共用;这两种藻类对甲基汞的吸附也可较好地由Freundlich等温吸附模型(R2 = 0.9248与0.8614)拟合[25],提示在藻细胞表面可能存在多个吸附位点同时起作用,且吸附位点间表现出不同的吸附自由能与剩余度.
通过对椭圆小球藻(Chlorella ellipticus)、鱼腥藻(Anabaena)、水华束丝藻和铜绿微囊藻的红外光谱研究,藻类通过细胞壁或胞外多聚物(extracellular polymeric substances,EPS)上广泛存在的氨基、巯基、羧基和羟基等官能团与甲基汞结合[35-36]. 不同官能团与甲基汞的结合机理不同,羟基和羧基等官能团通过孤对电子与甲基汞形成配位键[37],磺酸基等官能团则通过失去电子形成负电荷后的静电作用吸附甲基汞[38]. 与藻体内含量较低的巯基等含硫基团相比,羟基与羧基对藻类吸附甲基汞的贡献可能较大也更易体现[36,39]. 羟基与羧基的吸附过程为环境中的甲基汞-配体复合物(CH3Hg-L)与细胞表面的官能团(R)经配体交换形成新的络合物(CH3Hg-R),CH3Hg-L与CH3Hg-R的相对热力学稳定常数决定了络合速率,此外L与R的空间位阻也可能影响吸附的速率,如谷胱甘肽、N-乙酰-L-半胱氨酸和N-乙酰-青霉胺等较大尺寸或支链较多的硫醇配体结合甲基汞的程度小于半胱氨酸和巯基乙酸等小分子配体[23].
1.1.3 藻类对甲基汞的吸收机制
研究发现三磷酸腺苷(adenosine triphosphate,ATP)和转运蛋白参与了藻类对甲基汞的吸收,表明藻类可以通过主动运输富集甲基汞. 光系统Ⅰ抑制剂(甲基紫精)、光系统Ⅱ抑制剂(二氯酚二甲基脲)、避光与γ-辐射等减少ATP的措施可抑制藻类对甲基汞的吸收,证明藻类存在需要ATP的甲基汞吸收过程[40-41]. 尽管藻类的甲基汞转运蛋白尚未确定,但在仓鼠卵巢细胞(ATCC CCL-61)中,LAT1转运蛋白承担了甲基汞-半胱氨酸复合物(MeHg-L-cysteine)的运输功能[41]. 研究猜测MeHg-L-cysteine的结构类似于LAT1转运蛋白的底物蛋氨酸,从而导致甲基汞被错误运输[42]. LAT1蛋白过表达的CHO-k1细胞中MeHg-L-cysteine与蛋氨酸的竞争吸收现象进一步支持了上述猜测的合理性[41]. 而参与藻类主动吸收甲基汞途径的转运蛋白仍需进一步研究.
另外,藻类也可通过被动扩散吸收水体中的甲基汞. 如不同pH和氯离子浓度下,甲基汞的辛醇-水分配系数与威氏海链藻对甲基汞的吸收速率呈正相关关系(r = 0.75)[43],而且温度升高10 ℃对甲基汞吸收速率的影响(Q10)值仅为0.7,显著低于温度对主动运输吸收速率的影响(普遍为2—3)[40,44],表明甲基汞可通过被动扩散进入藻细胞. 但有研究发现经热灭活破坏主动运输途径后,梅尼小环藻(Cyclotella meneghiniana)的甲基汞吸收量显著下降,甲基汞在细胞质中占比由64%降至4%,证明被动扩散在甲基汞吸收中的贡献可能较低[45].
1.2 藻类对甲基汞的解吸与外排
藻类可能存在解吸与外排降低藻类中甲基汞的含量(图1). 在暴露168 h后,蛋白核小球藻的甲基汞吸附量相较最高吸附量(24 h)下降约40%,单个斜生栅藻的甲基汞吸收量相较最高吸收量(24 h)下降约74%[28]. 以上藻类对甲基汞的吸附和吸收的降低与藻类生长稀释以及增殖稀释有关[21],但也可能是由于甲基汞发生解吸和外排.
由于藻类对甲基汞的吸附、吸收、解吸和外排等过程可能同时发生,藻类对甲基汞的吸附和外排过程难以观察与定量. 藻类中甲基汞的解吸过程未见报道,但有研究表明震荡可使椭圆小球藻、水华束丝藻和铜绿微囊藻中69%、42%和34%的二价汞发生解吸[46]. 对于藻类中甲基汞的外排,过去研究认为甲基汞胁迫下藻类细胞膜通透性改变是原因之一[47]. 另外,甲基汞胁迫会导致莱茵衣藻(Chlamydomonas reinhardtii)中的铁转运蛋白、锌转运蛋白、ATP结合转运蛋白与阳离子扩散蛋白等金属转运相关蛋白质表达上调,因此,甲基汞外排可能需要转运蛋白的参与[26]. 但甲基汞胁迫下,一些蛋白质的高表达并非参与甲基汞的外排,如ATP结合蛋白的基因表达上调增加了胞内MRP2蛋白的含量,此蛋白可将甲基汞转移到液泡中而非排出体外[48]. 因此,后续需对甲基汞外排相关转运蛋白的具体作用机制进行研究.
1.3 藻类对汞甲基化和去甲基化的作用
目前,没有证据表明藻类可以直接产生甲基汞,但有研究认为藻类逆境中产生的代谢物二甲基硫分解为甲磺酸的过程可能导致甲基汞的生成[49]. 以往研究发现北极环境雪与地表水样品中的甲磺酸量与甲基汞量呈正相关[50],且二甲基硫转化过程中具有与硫还原菌相似的四氢叶酸途径[51],表明该途径可能存在,但暂未得到证实.
已有研究证据表明藻类可通过影响产甲基汞菌活性间接改变甲基汞浓度. 如向巴西采集的含微生物环境样品中加入蓝藻后,甲基汞净产率由6.8%增加至24.6%,而仅存在蓝藻的对照组水样中无甲基汞的产生[52]. 产甲基汞菌活性的提高可能是因为蓝藻增加了细菌群落所需的氢气[53],同时藻源性的叶绿素、蛋白质、细胞壁和脂质等物质也可增加产甲基汞细菌活性[54]. 另外,不同环境样品中蓝藻生物量与甲基汞含量呈正相关趋势[55]. 然而,藻类也可能抑制甲基汞产生,如中肋骨条藻(Skeletonema costatum)通过吸附二价汞(47%)降低了铁还原菌的甲基汞产量[56]. 此外,藻类也可能通过改变水体透光率以及溶解氧含量等影响汞的甲基化[57].
除光致去甲基化[58]与化学去甲基化[59]外,最近研究发现浮游生物群落也可在24 h内降解水样中12%的甲基汞[60]. 双富集同位素技术证明三角褐指藻(Phaeodactylum tricornutum)与旋链角毛藻(Chaetoceros curvisetus)等6种藻类可以引起甲基汞去甲基化,其中三角褐指藻等藻类可通过胞外分泌物实现甲基汞的光致去甲基化,东海原甲藻(Prorocentrum donghaiense)的去甲基化能力源于其伴生细菌,旋链角刺藻的去甲基化作用则来自于伴生细菌与胞外分泌物的共同作用[61]. 上述不同途径的去甲基化速率存在差异,胞外分泌物介导的光致去甲基化速率(0.01—0.39 d−1)显著高于伴生细菌的生物去甲基化(0.03—0.14 d−1)[61],该降解速率差异是由于反应机理不同[62-63]. 胞外分泌物被认为通过硫醇介导光致去甲基化[61],其可作为软碱与甲基汞的结合促进甲基汞的光降解[64];伴生细菌则可能通过甲基汞裂解酶(MerB)等途径降解甲基汞[65]. 但针对藻类及其伴生菌去甲基化过程的研究较少.
藻体内还存在其他改变环境中汞迁移性和生物可利用性的转化途径. 藻类可将二价汞还原为毒性低而挥发性强的溶解性零价汞[66],但此途径效率较低(< 5%),且机理尚不明确[67]. 通过冷原子荧光检测改进的酸碱还原差值法分析,硫化汞(β-HgS)也可能存在于藻类细胞中[68]. 有观点认为β-HgS是藻类中主要的无机汞形态(20%—90%),并推断其主要来源于液泡中二价汞的转化,但此前检测方法可能无法有效区分二价汞与硫化汞[69]. 未来可借助扩展X射线吸收精细结构谱等手段对硫化汞的生成过程进行详细研究[70].
1.4 藻类中甲基汞的亚细胞分布
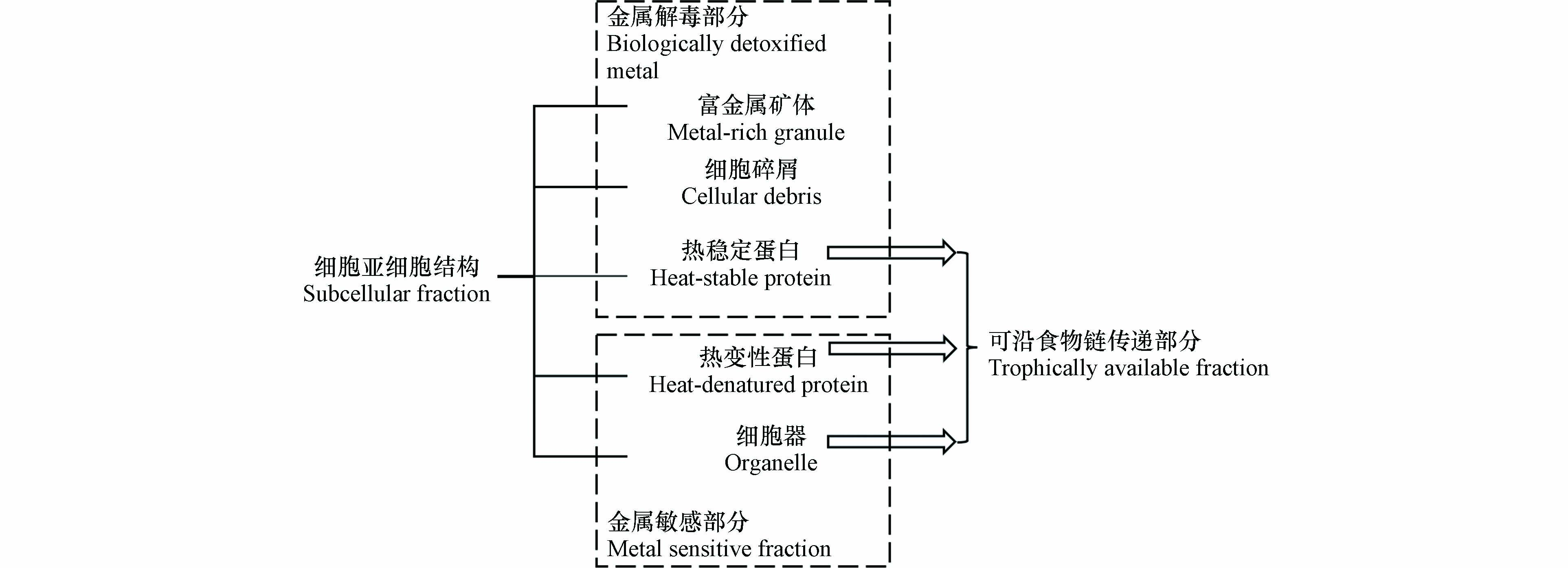
通过差速离心、热处理与化学处理可将藻类分为细胞碎屑(包括细胞壁与细胞膜等)、细胞器、热稳定蛋白(如植物螯合肽等)、热变性蛋白(如酶等)与富金属矿体5部分[71-72]. 通常将富金属矿体、细胞碎屑与热稳定蛋白统称为金属解毒部分即生物非活性部分,将细胞器与热变性蛋白划分为金属敏感组分. 热稳定蛋白是甲基汞在藻类中的主要结合部分,其中甲基汞的累积量可占细胞总累积量的44%(假微型海链藻)、68%(小球藻)和80%(等鞭金藻)[31]. 在热稳定蛋白中,甲基汞主要与含巯基的半胱氨酸、谷胱甘肽和植物螯合肽等组分络合[33,73]. 与二价汞类似,甲基汞在热稳定蛋白中达到饱和后,可能会进一步累积到其他结构中,因此细胞碎屑与细胞器组分中也存在甲基汞,但均未超过总累积量的30%[31].
藻类中甲基汞的亚细胞分布对其生物可利用性可能有重要影响[74]. 图2总结了不同累积位点重金属的生物可利用性差异,细胞器与热变性蛋白等金属敏感部分中镉与银等重金属可沿食物链传递,而细胞碎屑与富金属颗粒等解毒组分中的重金属则不会沿食物链传递[75-77]. 以往研究通过明胶包裹含甲基汞的贻贝不同亚细胞组分喂食鱼类发现,热稳定蛋白、热变性蛋白和细胞器等组分中甲基汞被同化的比例高于不溶物等其他组分[78]. 而分布于不同亚细胞结构的重金属在食物链传递中存在差异[79]. 目前尚未开展藻类富集甲基汞的相关研究,藻类不同亚细胞结构在甲基汞传递中的作用还需进一步研究.
1.5 影响藻类富集甲基汞的因素
藻类富集甲基汞受生物因素和环境因素影响. 藻类的相对表面积、结构以及活性均是影响富集效果的重要生物因素,环境因素则包括pH、温度、溶解性有机质(dissolved organic matter,DOM)、氯离子和硒等.
1.5.1 生物因素
藻类的相对表面积可影响藻类富集甲基汞的能力. 齿状藻等藻类甲基汞的VCF与相对表面积的相关系数可达0.97[34]. 因此,相对表面积更大的原核藻类蓝藻的甲基汞富集能力高于常见的硅藻与隐藻等真核藻类[47].
藻类结构也影响甲基汞的富集. 如没有叶绿体的原核藻类裂须藻的甲基汞富集率显著低于被内质网包裹叶绿体的真核藻类海链藻[80],且这两种藻类的甲基汞富集量均小于含有完整的脂质双分子层叶绿体的鼓藻与月牙藻(Selenastrum)[40],说明了叶绿体膜在转运和累积甲基汞可能发挥重要功能. 此外,藻类结构差异可能导致甲基汞的后续传递差异,如浮游动物与聚球藻以及原绿球藻的甲基汞浓度之比仅为0.7左右,显著低于硅藻等真核藻类,这可能因为原核细胞的细胞器较少,胞内膜结构不发达使甲基汞更趋于在藻类细胞碎屑等金属解毒部分累积,进而降低甲基汞在食物链中的传递效率(图2)[81].
藻类的细胞活性也是影响其富集甲基汞的重要因素. 稳定生长阶段藻类用于主动运输的能量较多[40],因此鼓藻在稳定生长阶段甲基汞吸收量(759 amol·cell−1)显著高于其在指数生长阶段的吸收量(38.1 amol·cell−1).
1.5.2 环境因素
(1)pH:藻类对重金属的吸收受pH调控. 以往研究认为pH下降时,甲基汞与H+竞争细胞表面的阳离子吸附位点引起藻类的甲基汞吸附量的降低[82],但目前证据显示pH下降增加藻类对甲基汞的富集. 研究发现全球范围内pH较低水体中藻类甲基汞含量普遍高于高pH水体[83]. 进一步的研究发现酸化增加藻类对甲基汞的富集,如当pH由6.5降低至5.5后,莱茵衣藻对氯化甲基汞的富集程度增加了1.6倍到2倍[84]. 这种现象可能与水体中甲基汞的赋存形态以及浓度改变有关:首先,酸化导致水体中与有机质结合的甲基汞释放,增加了具有生物可利用性的甲基汞量[85];其次,pH较低时,甲基汞的主要形态为更易进入细胞的氯化甲基汞,其生物可利用高于其他汞形态[43];另外,酸化条件下,细胞膜通透性会随藻类脂质与叶绿体改变而变化,促进甲基汞的扩散[86].
(2)温度:气候变暖已经成为全球面临的重要问题. 2010年全球海水表层平均温度较100年前升高了0.6 ℃[87]. 温度升高不仅引起海洋的酸化[88],也可促进藻类的生长繁殖,提高藻类生产力,进而影响藻类对甲基汞的富集[89]. 实验室条件下模拟实验显示温度由20 ℃增长至40 ℃时,羊角月牙藻的甲基汞吸收速率上升70%[40]. 但温度改变对单位生物量藻中甲基汞含量影响较小,提示升温可能通过增加藻类生物量促进其累积甲基汞[90]. 因此,虽然藻类生长繁殖受温度改变影响较大,藻类富集甲基汞的能力对温度变化并不敏感[21].
(3)溶解性有机质:多数情况下,DOM通过巯基结合甲基汞进而降低藻类对甲基汞的富集[91]. 高浓度DOM(20 mg·L−1)的添加可使梅尼小环藻甲基汞的VCF下降90%,低浓度的DOM(1.5 mg·L−1)也使其VCF下降一半[92]. 此外,疏水DOM对藻类富集甲基汞的抑制效果比亲水DOM更显著,这可能是由于疏水DOM中与甲基汞结合的双齿芳香基团的含量更高,比亲水DOM更易络合甲基汞[92].
但是,也有研究发现梅尼小环藻、莱茵衣藻和隐鞭藻在高DOM环境水体(旧金山湾三角洲)中甲基汞VCF是其在低DOM环境水体(科苏姆内斯河)中的2倍以上[45]. 该现象可能与DOM的异质性有关. 不同来源的DOM对藻类累积甲基汞的影响存在差异. 通过大肠杆菌生物传感器揭示DOM对甲基汞细胞富集的影响,结果为来自原绿藻和易变裸藻的EPS严重抑制了甲基汞的富集,而来自纤细裸藻的EPS的抑制效果并不显著[93]. 此外,蓝藻的EPS也在一定程度上降低铜绿假单胞菌(Microcystis aeruginosa)对甲基汞的富集[94],上述结果提示了不同来源的EPS可能对藻类富集甲基汞的影响效果存在差异. 通过分离和分析EPS中不同分子量的组分发现氨基酸与多胺的量与甲基汞富集量呈正相关,而羧基及其衍生物与甲基汞富集量呈负相关[93]. 对小分子DOM模型研究发现,不同DOM的形态与配位作用差异也影响了谷胱甘肽、半胱氨酸和巯基乙酸等含巯基化合物对藻类富集甲基汞的抑制效果[24].
(4)其他因素:研究发现水体中氯离子和硒也影响藻类累积甲基汞. 氯离子可与甲基汞络合,当氯离子浓度由0.47 mmol·L−1增加至470 mmol·L−1时,水体中氯化甲基汞的占比由13%增至99%,布氏双尾藻对甲基汞的VCF由4.0×104增至1.1×105[34],这可能是由于氯化甲基汞的膜渗透率高于氢氧化甲基汞[43]. 硒代甲硫氨酸可使海链藻对二价汞的4 h富集率由30%增长至70%,使甲基汞的4 h富集率从75%下降至44%[95]. 这可能是由于二价汞、甲基汞与硒代甲硫氨酸形成的复合物的跨膜特性不同[96],而胞内汞硒复合物的生成也可能影响藻类对甲基汞的吸收、外排及营养级传递[95].
2. 藻类与浮游动物间甲基汞传递特征(Trophic transfer characteristics of methylmercury bioaccumulated from algae to zooplankton)
浮游动物可通过摄食藻类进而累积汞,其对甲基汞与二价汞的同化效率存在显著差异[20,97]. 如汤氏纺锤水溞(Acartia tonsa)对藻类饵料中甲基汞的同化效率为58%—79%,对二价汞的同化效率仅为25%—31%,导致甲基汞的营养级放大因子(> 1)显著高于二价汞(0.2)[98].
甲基汞营养级传递受藻类和浮游动物等生物因素影响. 研究表明藻类细胞质中甲基汞含量与浮游动物的同化率有密切关系(r = 0.95),提示细胞质中甲基汞更易被浮游动物同化[98]. 另外,藻类种类也影响甲基汞在浮游动物中的同化作用,如汤氏纺锤水溞对小型假微型海链藻甲基汞的同化率(71%)低于同属大体积的威氏海链藻的同化率(88%)[99]. 因此,不同藻类对食物链甲基汞传递的贡献不同,模型结果表明硅藻与聚球藻分别贡献了沿食物链转移甲基汞的35%与25%,而其余藻类对甲基汞的贡献为40%[81]. 此外,浮游动物的甲基汞浓度随其体积增加而增加[100],甲基汞同化率随着肠道通过时间的增加而增加[101].
环境变化也是影响甲基汞食物链传递的重要因素[102]. 升温可在一定程度上增加产甲基汞菌的甲基化活性[103]以及藻类对甲基汞的累积[100],也可通过改变藻类消费者的生命活动(如排泄与生长速率等)影响甲基汞的食物链传递[90]. 当从14 ℃增至24 ℃时,大型溞对藻类的甲基汞同化率无显著变化,但显著影响排泄和生殖过程对大型溞体内甲基汞排出量的贡献,其中排泄的贡献率由52%升至85%,生殖的贡献率由43%降至11%,总外排量降低使甲基汞累积增加[104]. 因此,温度变化可通过改变浮游动物生命活动来影响甲基汞食物链传递.
目前广泛发生的富营养化也影响甲基汞的食物链传递,但是具体效果存在争议. 一种观点认为富营养化可以增加藻类生物量,降低单个藻类甲基汞的累积,如富营养化使藻类生物量增加3倍时,浮游植物甲基汞含量降低,进而导致水溞的甲基汞含量下降70%[105]. 另一种观点认为富营养化可提高水体中甲基汞的浓度. 富营养化引起的藻源性有机质的增加可刺激微生物汞甲基化[106],藻类的大规模凋亡阶段也会释放大量甲基汞[107]. 模型结果表明富营养化可使波罗的海水体内甲基汞总量增加4倍,间接增加藻类对甲基汞的富集[106]. 在巢湖、东湖与滇池水样品中加入藻类可使甲基汞产量提升了24.3%—15918%[108]. 但是,富营养化对甲基汞食物链传递的综合影响尚需深入探究.
3. 总结与展望(Conclusions and perspectives)
藻类可通过吸附、主动运输和被动扩散富集水中的甲基汞,并通过转化、解吸、外排和食物链传递等过程影响甲基汞的环境归趋. 生物与环境因素是影响甲基汞藻类累积及其食物链传递的重要因素. 由于藻类种类繁多且生理状态多变,不同区域pH、温度和DOM等环境条件差异巨大,因此藻类富集和传递甲基汞过程非常复杂. 目前,藻类富集甲基汞的机制与影响因素尚待厘清. 如藻类主动吸收甲基汞的转运蛋白与机制仍不清楚,藻类对甲基汞的主动外排和转化途径(如甲基化与硫化)仍有待证实. 此外,pH和DOM等环境因素对藻类富集甲基汞与后续食物链传递的影响存在争议,需深入研究.
目前,藻类富集甲基汞及其食物链传递的研究大部分为实验室模拟实验,未来可将实验室模拟实验与长期的现场研究相结合,以明确实际环境中甲基汞的藻类累积和食物链传递行为. 多同位素示踪、全细胞生物传感器和同步辐射等技术可望在阐明酸化、富营养化和全球变暖等背景下藻类对甲基汞富集与后续营养传递中扮演重要角色.
-
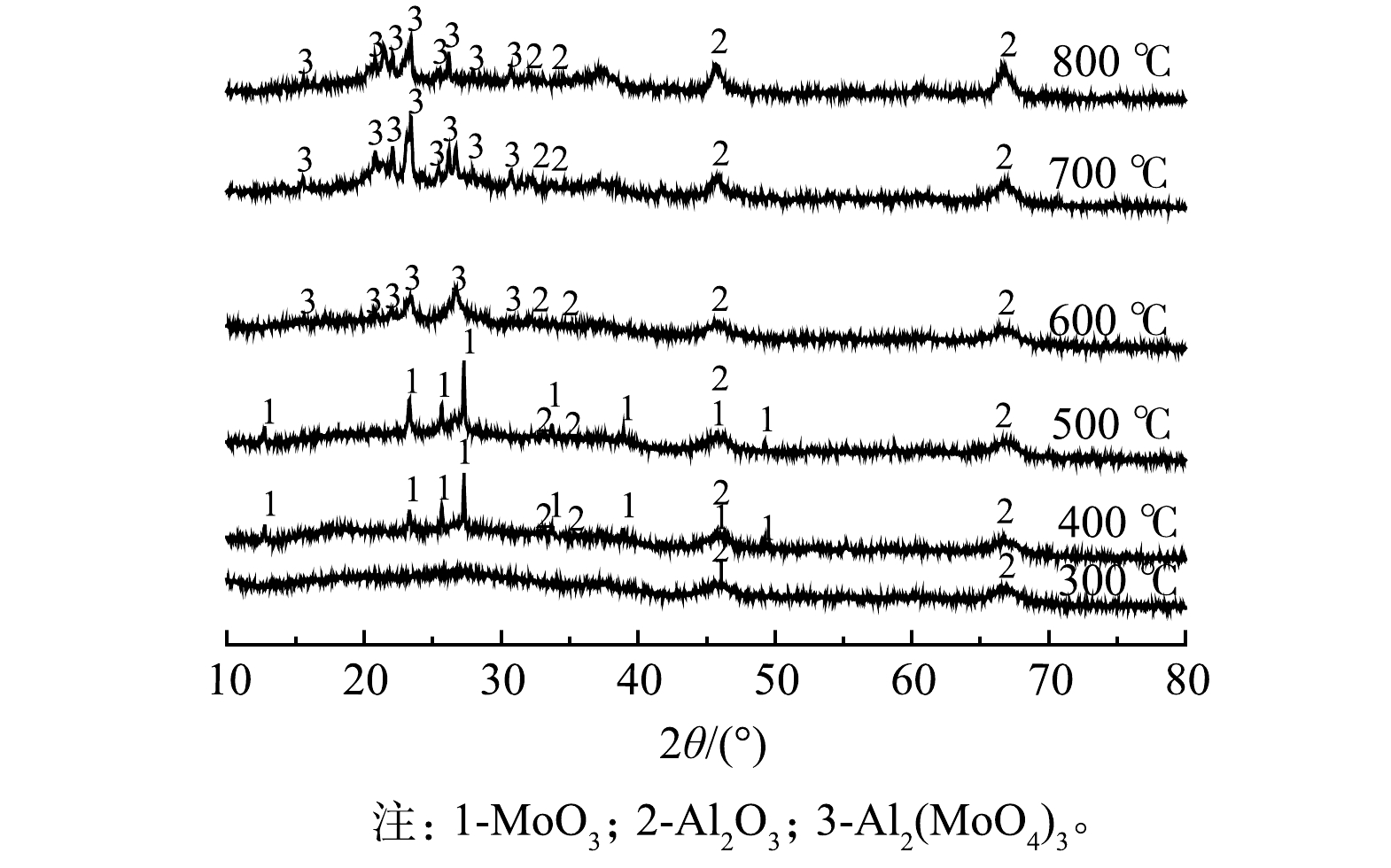
图 1 不同预处理温度下废催化剂物相分析
Figure 1. Phase analysis of spent catalyst at different pretreatment temperatures
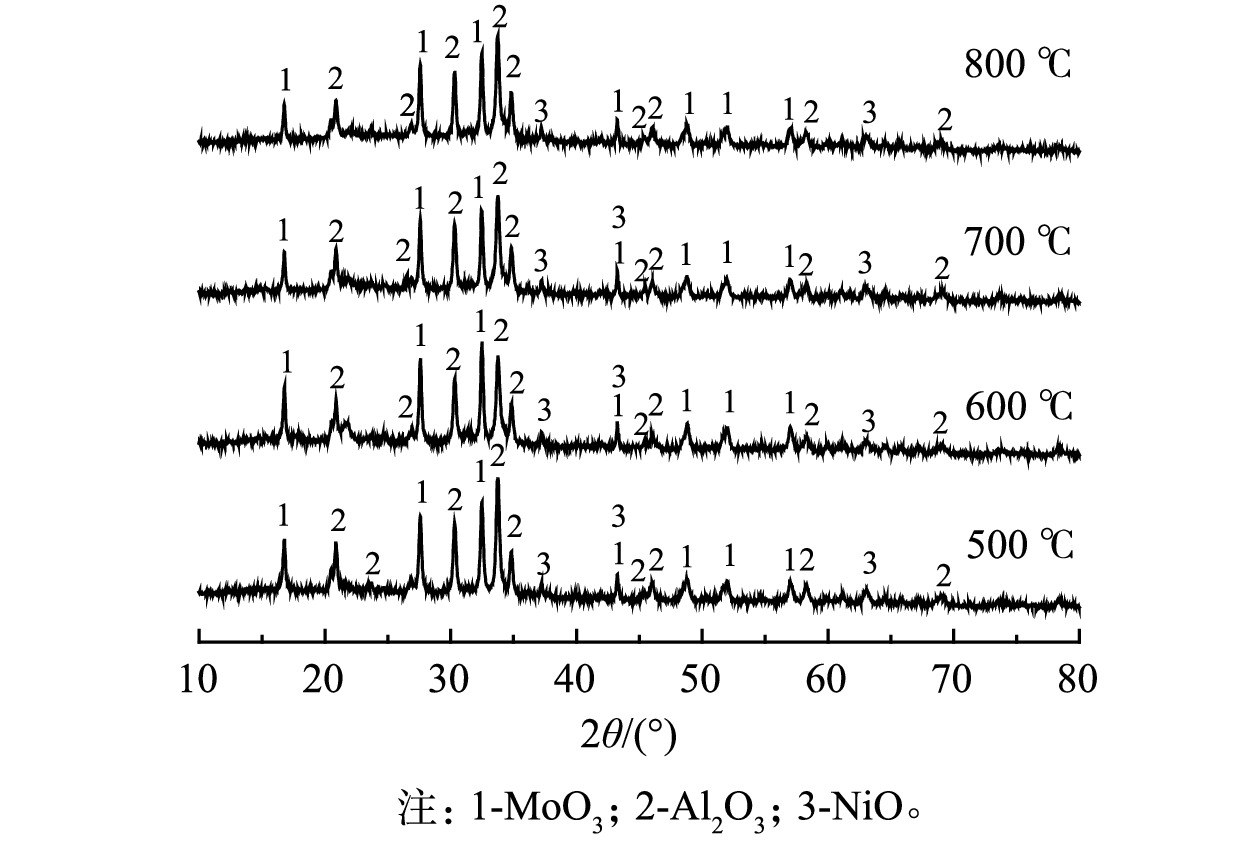
图 2 不同温度下预处理废催化剂钠化反应后产物物相分析
Figure 2. Phase analysis of sodium reaction of waste catalyst pretreated at different temperatures
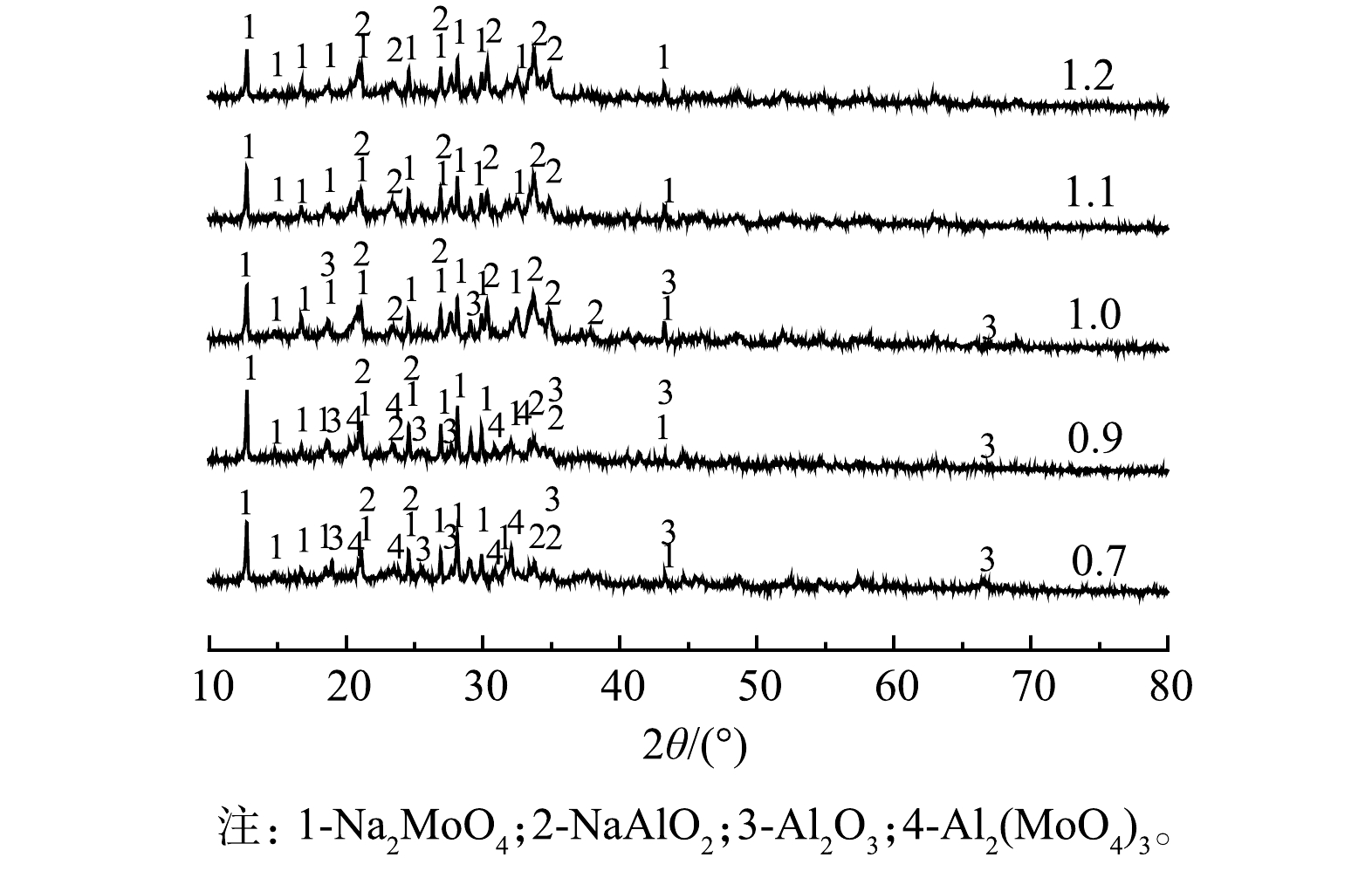
图 4 添加不同量碳酸钠的焙烧产物 (950 ℃) 物相分析
Figure 4. Phase analysis of calcined products (950 ℃) with different amount of sodium carbonate
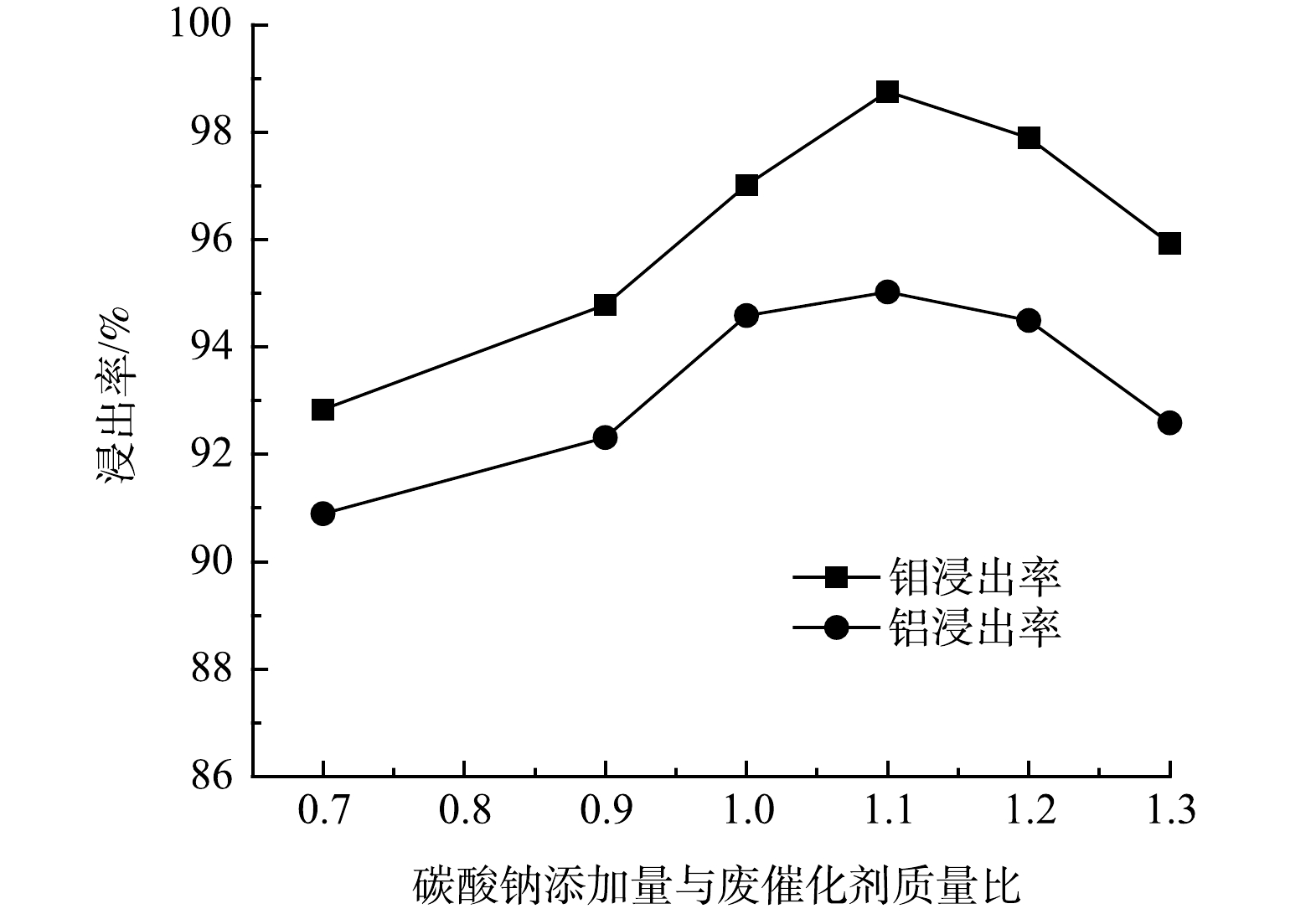
图 5 不同碳酸钠添加量下Mo、Al浸出率
Figure 5. Leaching rate of molybdenum and aluminum with different amount of sodium carbonate
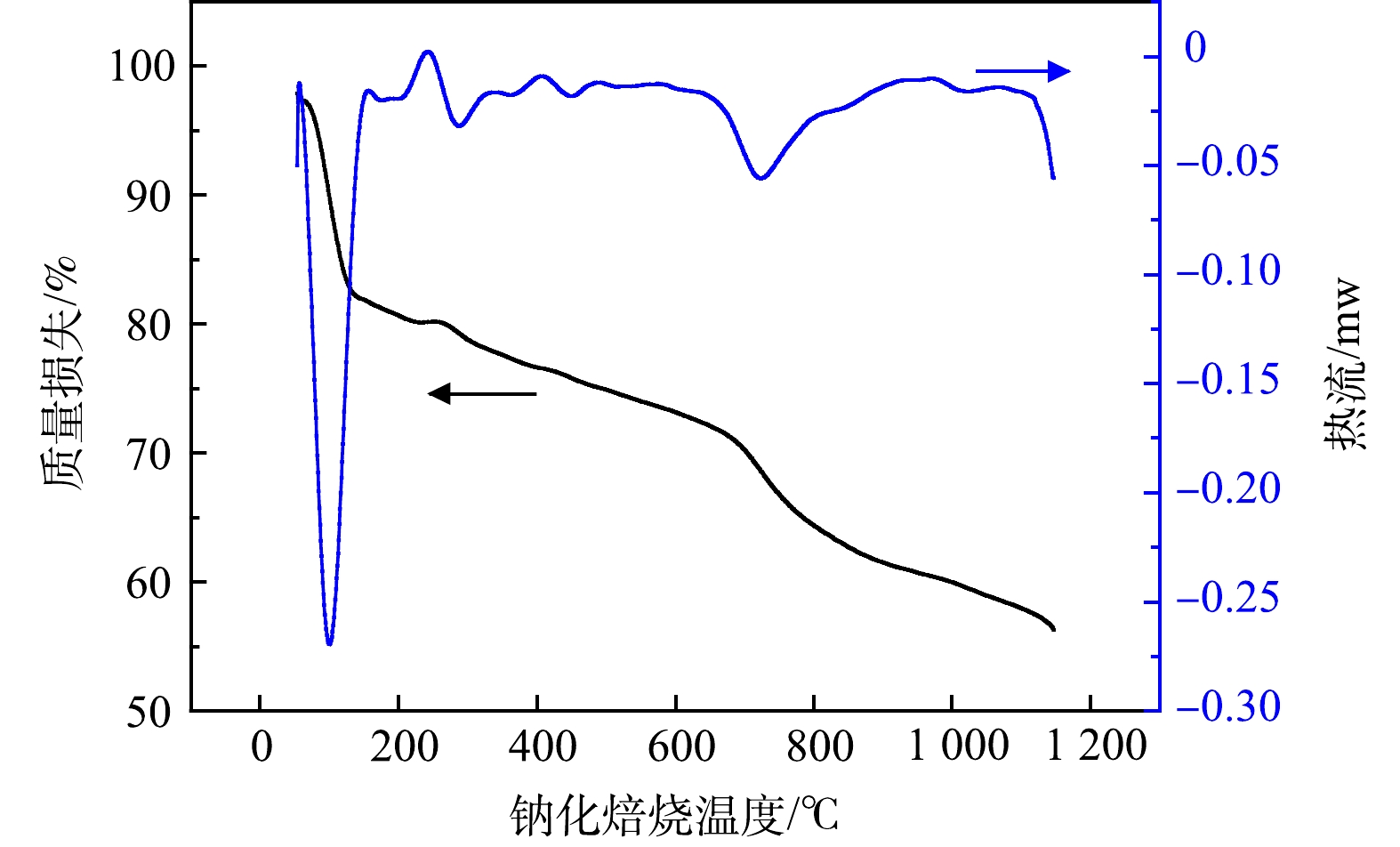
图 6 预处理废加氢催化剂与碳酸钠混合物的TG-DSC曲线
Figure 6. TG-DSC curves of the mixture of pretreated spent hydrotreating catalyst and sodium carbonate
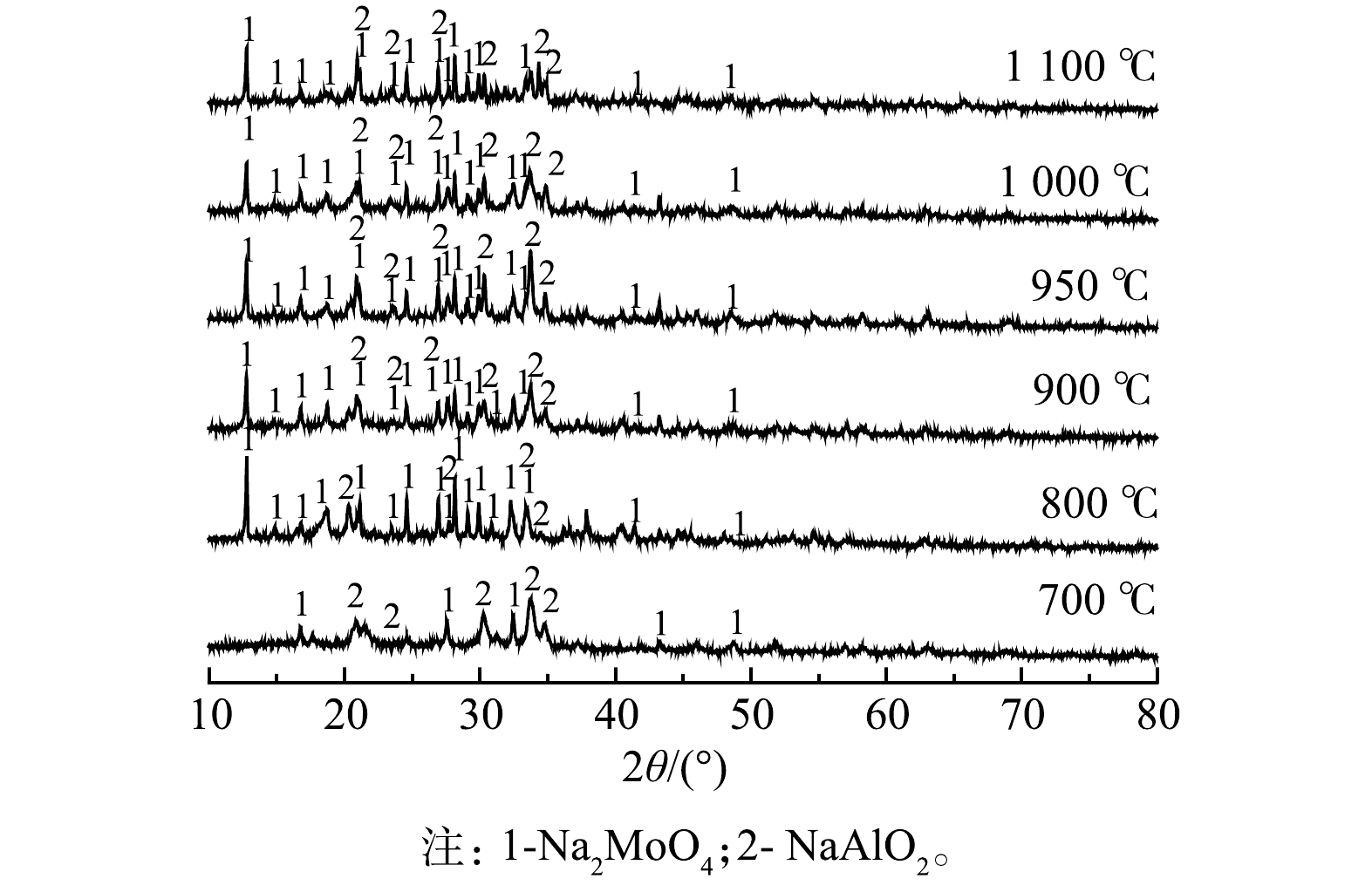
图 7 不同焙烧温度下产物的物相分析
Figure 7. Phase analysis of products at different calcination temperatures
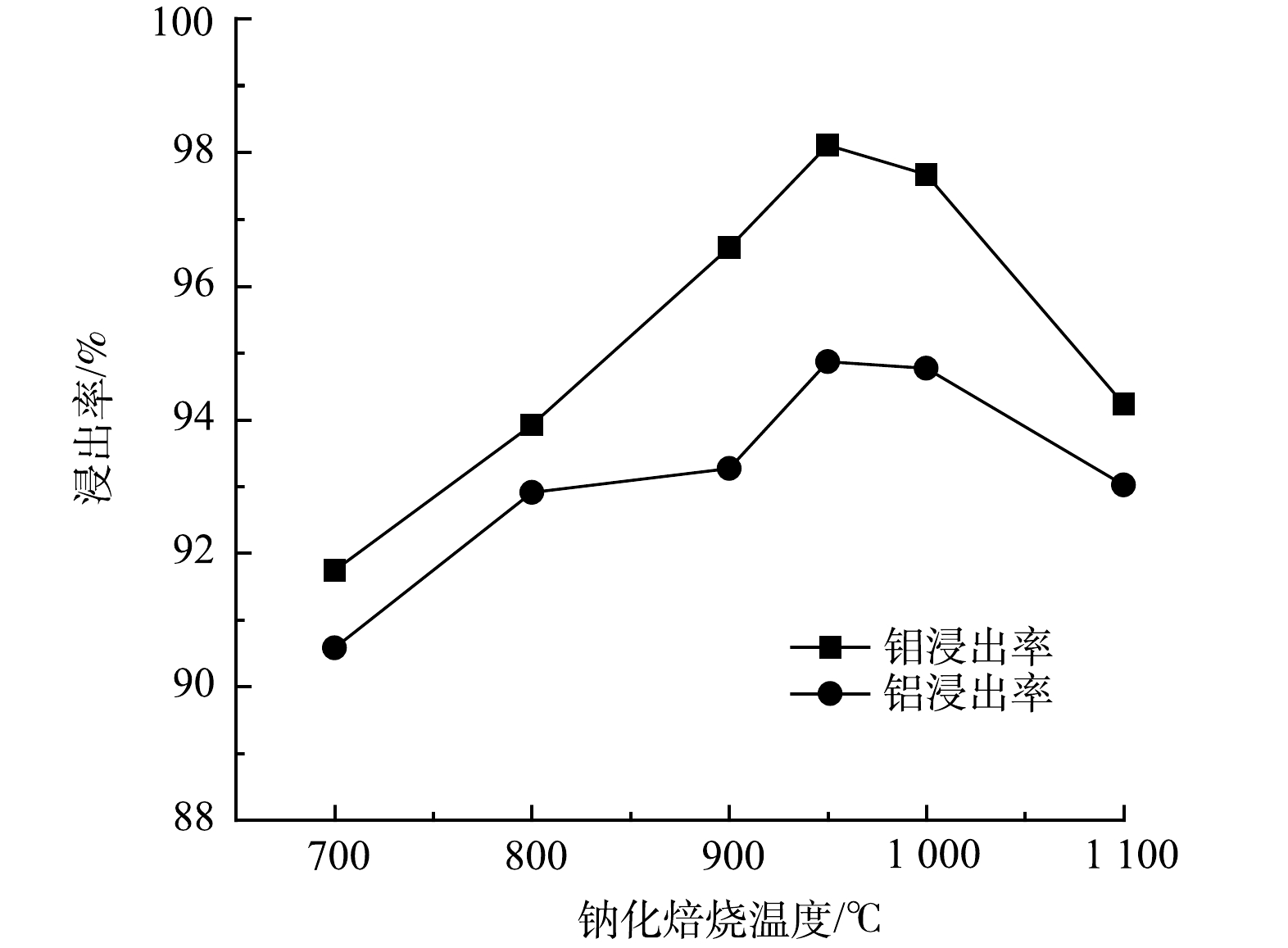
图 8 不同钠化焙烧温度下Mo、Al浸出率
Figure 8. Leaching rate of molybdenum under different roasting temperature and sodium
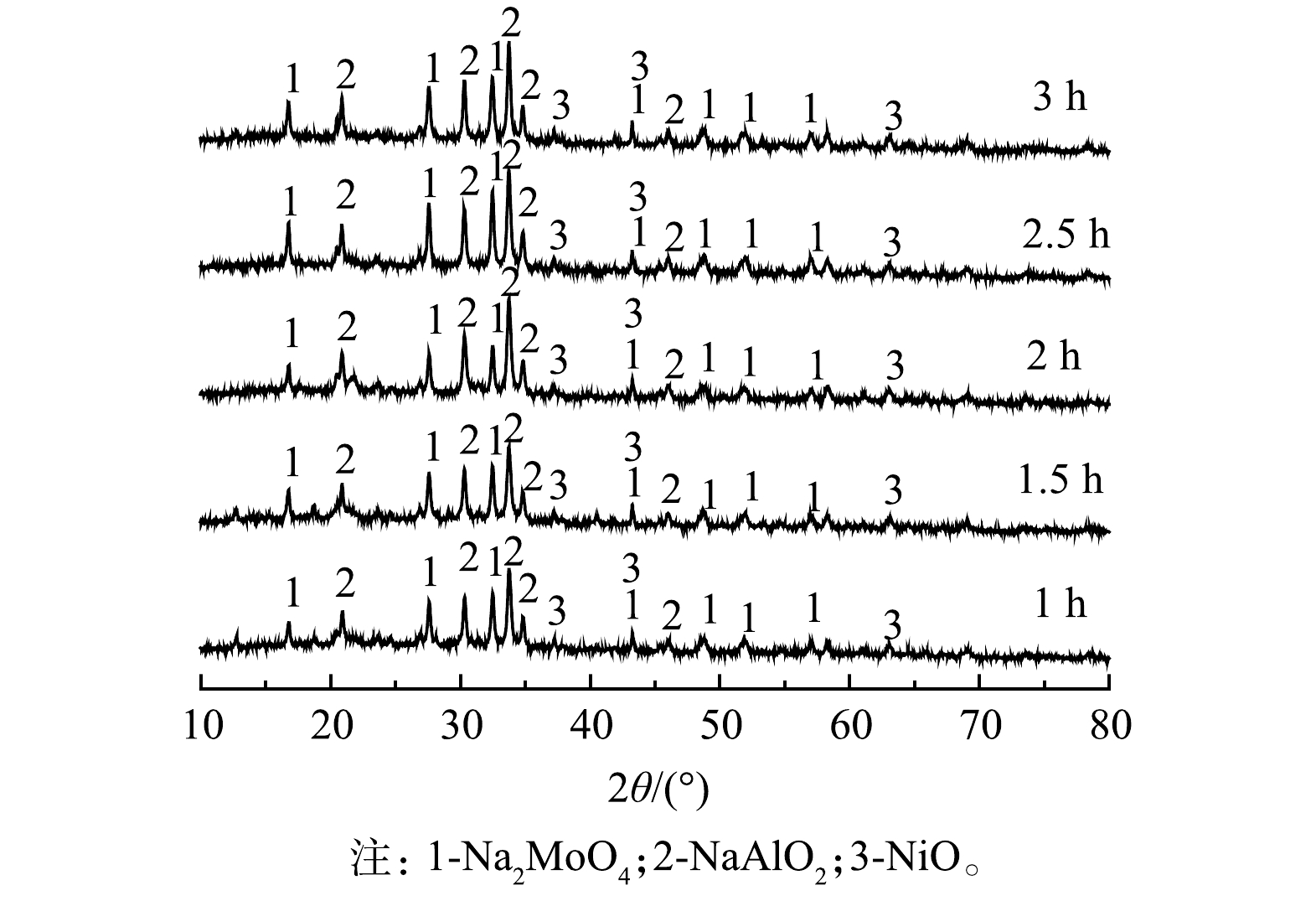
图 9 不同焙烧时间条件下焙烧产物 (950 ℃) 的物相分析
Figure 9. Phase analysis of calcined products (950 ℃) under different calcination time
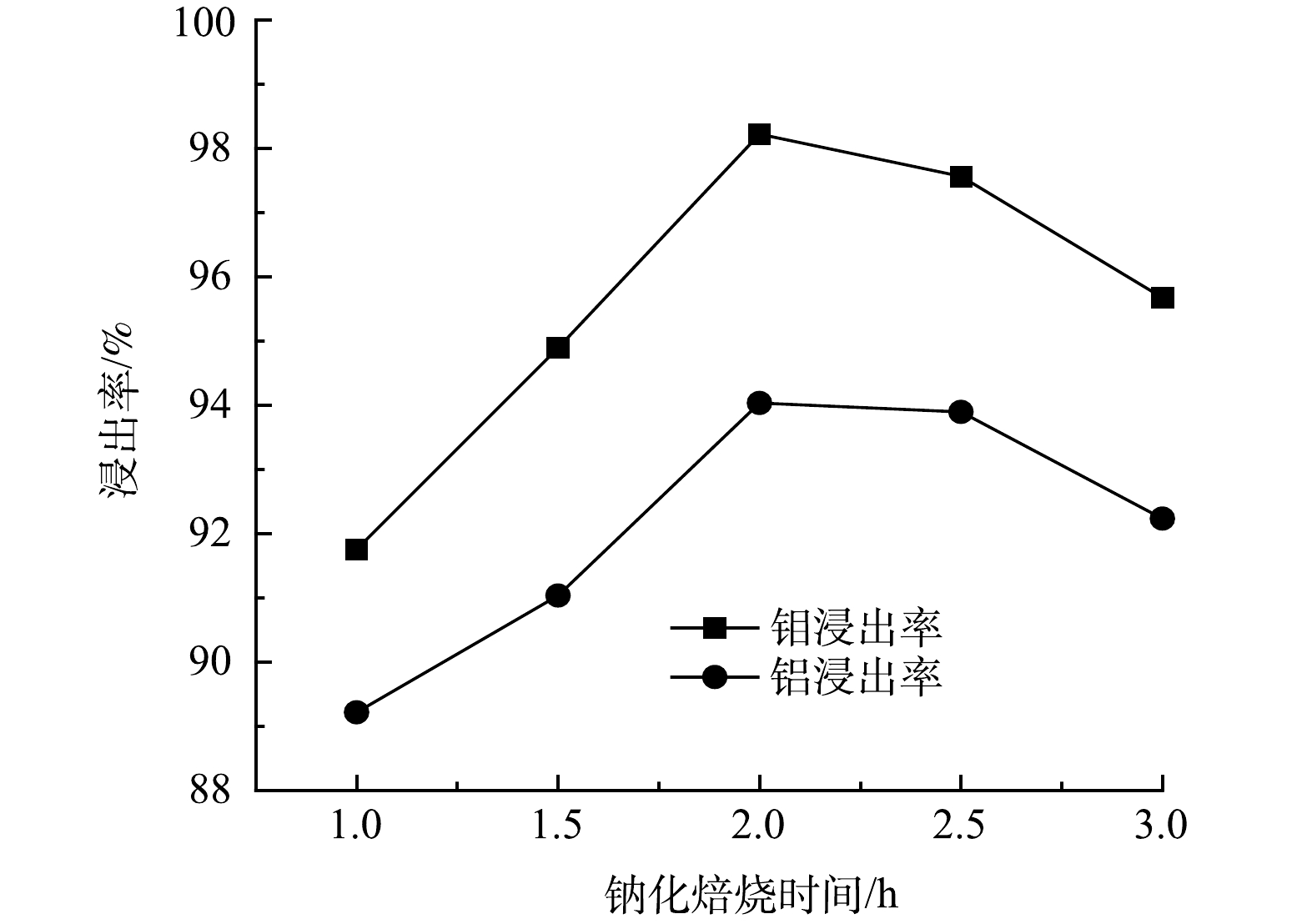
图 10 不同钠化焙烧时间下Mo、Al浸出率
Figure 10. Leaching rate of molybdenum and aluminum under different sodium roasting time
表 1 废催化剂XRF分析结果
Table 1. Main composition of the material by XRF %
Al2O3 MoO3 SO3 P2O5 NiO SiO2 Fe2O3 CoO 47.82 21.71 20.25 4.317 3.558 2.08 0.072 9 0.071 8
下载: 导出CSV
表 2 BBD法实验设计
Table 2. Design experiment level and coding with BBD method
因素 水平参数 −1 0 +1 焙烧温度(A)/ ℃ 800 900 1100 焙烧时间(B)/h 1 2 3 碳酸钠添加量(C) 0.7 1.1 1.5 浸出液固比(D) 3∶1 6∶1 9∶1 浸出温度(E)/ ℃ 40 60 80
下载: 导出CSV
表 3 废催化剂在不同温度下的烧矢量
Table 3. Burning vector of spent catalyst at different temperatures
温度/ ℃ 烧失量/% 温度/ ℃ 烧失量/% 300 10.2 650 19.2 400 17.4 700 19.6 500 17.9 800 21.5 600 18.7
下载: 导出CSV
表 4 回归模型的方差分析结果
Table 4. Analysis of variance of regression model
参数 自由度 平方和 均方差 F值 P Mo Al Mo Al Mo Al Mo Al Model 20 605.72 757.30 30.29 37.86 15.97 10.00 <0.000 1 <0.000 1 A 1 44.02 40.10 44.02 40.10 23.21 10.59 <0.000 1 0.003 3 B 1 250.27 310.64 250.27 310.64 131.96 82.04 <0.000 1 <0.000 1 C 1 1.06 0.55 1.06 0.55 0.56 0.14 0.001 5 0.006 9 D 1 0.028 0.56 0.028 0.56 0.015 0.15 0.904 2 0.704 1 E 1 10.26 5.69 10.26 5.69 5.41 1.5 0.028 5 0.231 7 AB 1 0.004 9 1.56 0.004 9 1.56 0.002 584 0.41 0.959 9 0.526 5 AC 1 1.31 2.89 1.31 2.89 0.69 076 0.413 6 0.390 6 AD 1 1.21 0.026 1.21 0.026 0.64 0.006 761 0.432 0 0.935 1 AE 1 0.016 2.74 0.016 2.74 0.008 283 0.72 0.928 4 0.403 1 BC 1 4.20 3.33 4.20 3.33 2.22 0.88 0.149 1 0.357 3 BD 1 1.00 2.03 1.00 2.03 0.53 0.54 0.474 5 0.470 8 BE 1 0.20 0.74 0.20 0.74 0.11 0.20 0.746 6 0.662 3 CD 1 0.21 0.58 0.21 0.58 0.11 0.15 0.741 2 0.699 4 CE 1 0.25 0.11 0.25 0.11 0.13 0.030 0.722 3 0.864 7 DE 1 0.018 0.023 0.018 0.023 0.009 609 0.005 942 0.922 7 0.939 2 A2 1 49.30 54.58 49.30 54.58 25.99 14.42 <0.000 1 0.000 8 B2 1 142.56 185.74 142.56 185.74 75.17 49.02 <0.000 1 <0.000 1 C2 1 189.11 243.46 189.11 243.46 99.71 64.30 <0.000 1 <0.000 1 D2 1 80.88 89.04 80.88 89.04 42.64 23.52 <0.000 1 <0.000 1 E2 1 7.13 2.11 7.13 2.11 3.76 0.56 <0.000 1 0.462 4 残差 25 47.42 94.66 1.90 — — — — — 失拟项 20 41.18 86.47 2.06 — 1.65 2.64 0.303 8 0.142 7 纯误差 5 6.24 8.19 1.25 — — — — — 总离差 45 653.13 851.96 — — — — — —
下载: 导出CSV
表 5 实验结果验证
Table 5. Verification of experimental results
% 实验序号 Mo浸出率 Al浸出率 1 98.79 95.21 2 99.01 94.89 3 99.24 95.03
下载: 导出CSV
-
[1] ISABEL Pinto, HELENA Soares. Recovery of molybdates from an alkaline leachate of spent hydrodesulphurisation catalyst – proposal of a nearly-closed process[J]. Journal of Cleaner Production, 2013, 52: 481-487. doi: 10.1016/j.jclepro.2013.03.021 [2] 刘公召, 阎伟, 梅晓丹, 等. 从废加氢催化剂中提取钼的研究[J]. 矿冶工程, 2010, 30(2): 70-72. doi: 10.3969/j.issn.0253-6099.2010.02.018 [3] 季思伟. 炼油加氢废催化剂中有价金属的回收[D]. 上海: 华东理工大学, 2012. [4] 许傲云. 石油加氢废催化剂中有价金属的综合回收利用[D]. 上海: 东华大学, 2014. [5] 赵中伟, 李江涛. 中国钼镍矿开发利用的技术现状及前景分析[J]. 中国金属通报, 2010(29): 38-40. [6] 张沛. 660MW燃煤电厂商用SCR脱硝催化剂的失活分析与再生探究[D]. 杭州: 浙江大学, 2017. [7] 祁兴维, 林爽. 废加氢催化剂中金属钼回收技术研究[J]. 当代化工, 2019, 48(04): 126-128+141. doi: 10.3969/j.issn.1671-0460.2019.04.030 [8] 刘贵清, 张邦胜, 王芳, 等. 废镍钼催化剂两段焙烧-水浸试验研究[J]. 中国资源综合利用, 2018(6): 23-26. doi: 10.3969/j.issn.1008-9500.2018.06.007 [9] 北京矿冶研究总院测试研究所. 有色冶金分析手册[J]. 北京:冶金工业出版社, 2004: 202-203. [10] 宋生强. 氧化钼、氧化钒自还原直接合金化冶炼含钼含钒钢研究[D]. 武汉: 武汉科技大学, 2014. [11] 向铁根. 钼冶金[J]. 长沙:中南大学出版社, 2009: 181-183. [12] 淡玄玄, 李小敏. 亚麻对染料甲基紫吸附的响应面法分析[J]. 环境监测管理与技术, 2017, 29(6): 68-71. doi: 10.3969/j.issn.1006-2009.2017.06.017 [13] 王雅辉, 邹雪刚, 舒冉君, 等. 胡敏素对Pb2+吸附的响应面优化及机理[J]. 中国环境科学, 2017, 37(5): 1814-1822. doi: 10.3969/j.issn.1000-6923.2017.05.026 [14] MOHAMED OSMAN Saeed, KHAIRUM Azizli, MOHAMMED HASNAIN Isa, et al. Application of CCD in RSM to obtain optimize treatment of POME using Fenton oxidation process[J]. Journal of Water Process Engineering, 2015, 52: 481-487. 期刊类型引用(4)
1. 曹耀华,彭大源,张勤,刘源,杨洪英. 响应曲面法优化废铂催化剂中难溶颗粒HCl-DCEA体系回收工艺. 中国有色冶金. 2024(03): 37-45 .  百度学术
百度学术
2. 邓颖,关洪亮. 废旧加氢催化剂中钼回收工艺的研究进展. 云南化工. 2024(10): 54-57 . 百度学术
3. 巴仲凯,张丙然,陈天友. 石油炼化加氢催化剂中贵金属回收工艺的效率与成本评估. 山西化工. 2024(11): 156-158+196 . 百度学术
4. 胡红辉,金浩哲,李国良,曹春梅. 超声波浸渍-凝胶法合成技术在制备悬浮床加氢催化剂中的应用分析. 当代化工. 2023(09): 2017-2021+2029 . 百度学术
其他类型引用(4)
-
 点击查看大图
点击查看大图
计量
- 文章访问数: 3923
- HTML全文浏览数: 3923
- PDF下载数: 154
- 施引文献: 8
