

-
在生物难降解有机污染物的处理中,类芬顿高级氧化(advanced oxidation process, AOP)技术受到广泛的关注[1]。其中以过硫酸盐高级氧化技术最为典型[2]。大量研究表明,过硫酸盐通过加热[3-4]、紫外[4]和过渡金属离子[5-6]等形式的活化,可以有效实现对有机物的降解。然而,常见的活化方法在实际应用中存在局限性。其中,热活化和紫外活化需要消耗额外的能量,而过渡金属离子的引入会导致化学污泥的产生以及潜在的二次污染。
近几年,各类型碳材料由于来源广泛、价格低廉、耐用性好、无化学污泥等优点逐渐受到关注,对其催化性能的研究也有许多报道[7-9]。通常,具有完美六角蜂窝拓扑结构的碳催化剂(石墨)常表现出较低的催化活性。值得注意的是,除六角蜂窝拓扑结构以外,碳材料中还存在缺陷结构,包括边缘缺陷、空位/空穴缺陷、拓扑缺陷等[10]。其中,边缘缺陷是指碎片状微小碳面的边界,是含量最丰富的缺陷类型[11-12]。边缘位置的碳原子通常具有高于面内碳原子的电荷密度,具有更高的电子活性。空位/空穴缺陷是指面内一个或多个碳原子的缺失,可认为是位于催化剂内部的边缘缺陷[13]。此外,碳面中常见的非六元环,如五元环、八元环等,是极为重要的拓扑缺陷。各类型拓扑缺陷中不规则的碳原子数通常会导致电子重排,使其具有良好的电子活性[14]。有研究[15-17]表明,富缺陷的碳催化剂比无缺陷碳催化剂具有更好的吸附和催化性能。
由于氮元素的电负性高于碳元素,将氮原子掺杂到碳材料中会显著破坏原有π共轭结构,造成局部电子重排[18-20],通常称为氮杂质缺陷。常见的氮掺杂方法分为原位合成法和后处理法。原位合成法可分为活化法、水热法、化学气相沉积法、模板法、溶胶-凝胶法等;而后处理法是指将氮源在一定条件下引入到已有的碳材料中,属于间接合成法。一般情况下,以氨气、氨水、尿素等氮源和碳源混合,通过高温热处理即可有效实现碳材料的氮掺杂,掺杂态的氮一般以吡咯氮、吡啶氮和石墨氮等形式存在。有研究表明,除了氮掺杂含量,氮杂质缺陷的种类也可能影响催化剂的催化活性[21]。氮缺陷的存在有利于过一硫酸盐(peroxymonosulfate,PMS)的吸附,并可通过与相邻碳原子的协同作用实现对PMS的高效活化。尽管已经开展了大量的研究,但对PMS的催化效率仍有待提高。此外,氮掺杂碳催化剂的催化性能与氮含量或缺陷种类之间的内在催化机制尚不清楚。
基于上述研究结果,本研究在氮气气氛下,使用一步热处理法制备了一系列不同氮含量的碳催化剂,分别通过场发射扫描电镜、高分辨透射电镜、X射线衍射分析、X射线光电子能谱等手段对所制备催化剂的物化性质进行了详细分析。将所制备催化剂与PMS结合,构建了非均相高级氧化体系用于双酚A(bisphenol A,BPA)的降解。对催化剂投加量、氧化剂投加量、初始pH、共存化学组分等条件的影响进行了详细分析,并通过顺磁共振分析、活性物种淬灭实验、电化学分析以及量子化学计算等手段,对可能的降解机理进行了深入分析。
-
二氰二胺(C2H4N4)、碳酸钾(K2CO3)、过硫酸氢钾复合盐(Oxone, KHSO5· 0.5KHSO5· 0.5K2SO4)、双酚A(BPA)、盐酸(HCl)、硝酸(HNO3)、氢氧化钠(NaOH)、叔丁醇(TBA)等试剂均购于上海麦克林公司(分析纯),无水乙醇、甲醇、乙腈等色谱纯有机溶剂均购于赛默飞公司,实验用水均出自Milli-Q超纯水系统。
-
1)催化剂的制备。将碳黑(CB)置于管式炉中,由室温开始,在N2保护下以10 ℃·min−1的升温速率加热至900 ℃,并在900 ℃保持2 h。冷却至室温后,通过收集固体可以得到CB900。将CB900(35 g)和二氰二胺(35 g)均匀混合置于管式炉中,由室温开始,在N2保护下以10 ℃·min−1的升温速率分别加热至600、700、800、900和1 000 ℃,并在达到目标温度后保持1 h。冷却至室温后,收集剩余固体,用1 mol·L−1硫酸在80 ℃的水浴加热下清洗2 h,然后用去离子水洗涤至中性。洗涤后的固体使用电热鼓风干燥箱在60 ℃干燥24 h,可以得到CN600、CN700、CN800、CN900和CN1000(定义为CNx,x为制备时恒温段温度)。
2)实验仪器。Quanta FEG 250场发射扫描电子显微镜(SEM)与FEI Tecnai G2 F20型透射电镜(TEM),用于表征催化剂的微观形貌;Rigaku Ultimate IV型X 射线衍射仪(XRD),用于催化剂晶相分析;Thermo Fisher K Alpha型X射线光电子能谱(XPS),用于催化剂反应前后组分价态变化分析;V-sorb 2800P型孔径分析仪,用于催化剂比表面积分析;Bruker EMXmicro-6/1电子顺磁共振(EPR),用于活性氧物种鉴别;Nicolet iS50型傅里叶红外光谱仪(ATR-FTIR),用于PMS在催化剂表面的吸附行为分析。电化学工作站(CHI660E),用于PMS活化过程的电化学行为分析,采用三电极体系,银-氯化银电极作为参比电极,铂电极作为对电极,碳玻电极作为工作电极,电解质采用100 mmol·L−1硫酸钠溶液。
3) BPA降解实验。使用HNO3(0.1 mol·L−1)或NaOH(0.1 mol·L−1)将BPA溶液pH调至6.5±0.1,而后将催化剂加入到BPA溶液中,通过超声振荡10~15 s,使催化剂均匀分散,然后向溶液中加入PMS以激活氧化反应。反应过程中采用磁力搅拌器以120 r·min−1转速搅拌,在不同时间点取水样1 mL,使用0.22 μm滤膜过滤样品后加入1.5 mL高效液相色谱样品瓶中,并立即用0.5 mL甲醇进行淬灭。在常规实验中,反应过程均在(25±0.5) ℃的水浴中完成,BPA浓度为40 μmol·L−1,PMS浓度为0.4 mmol·L−1,催化剂投加量为0.1 g·L−1,初始pH为6.5±0.1。所取水样在2 h内使用超高效液相色谱进行测定,流动相采用水和乙腈混合液(乙腈体积比为40%),流速为0.2 mL·min−1,检测波长为278 nm,柱温控制为35 ℃。表观降解动力学常数根据式(1)进行计算。
式中:C0为BPA初始浓度,μmol·L−1;Ct为t时刻BPA浓度,μmol·L−1;t为时间,min;kapp为表观动力学常数,min−1。
-
通过SEM对所制备的催化剂的形貌进行了表征。由图1(a)~(e)可以看出,所制备的CNx催化剂颗粒尺寸分序差别大,但随着制备温度的升高,其平均颗粒尺寸逐渐变小。HR-TEM图像如图1(f)所示,可以发现CNx为短程有序状态,晶化度较低。其晶面间距为0.344 nm,与石墨的特征间距接近。
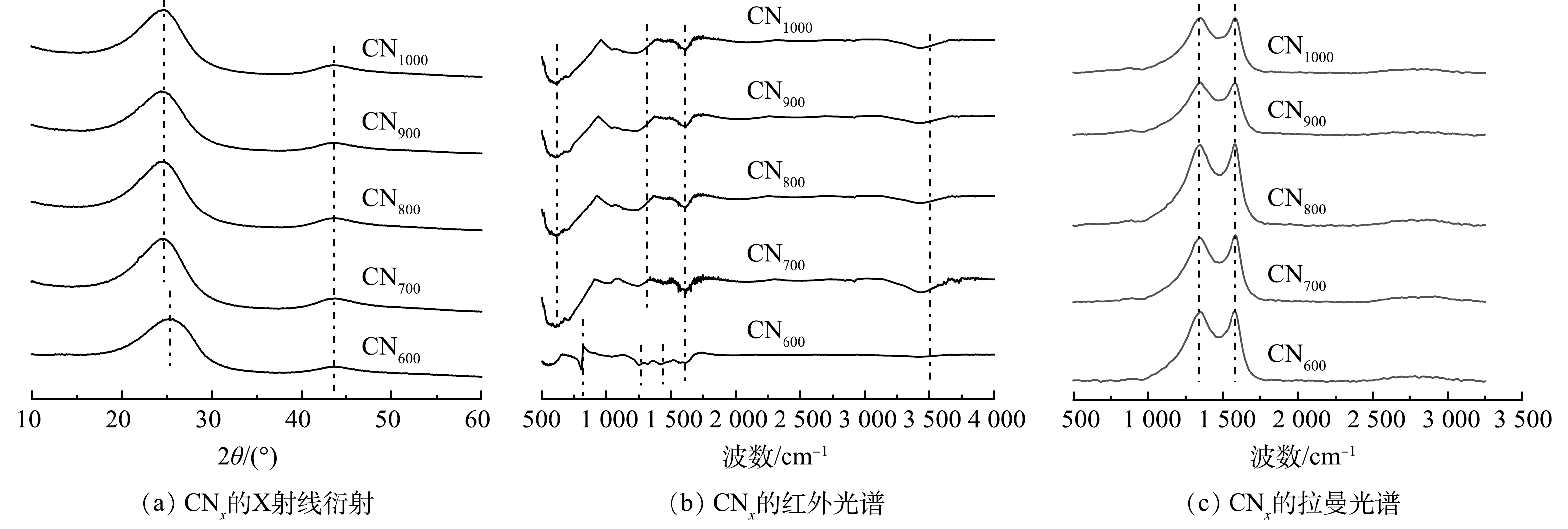
使用XRD分析对催化剂的物相信息进行进一步确定,结果如图2(a)所示。所制备的催化剂在24.6 °和43.6 °处存在2个衍射峰,分别对应于石墨的(002)和(004)面,且不同温度下制备的催化剂物相结构无显著变化。图2(b)为CNx的FT-IR光谱分析图。在3 417 cm−1处的宽峰对应N—H、—COOH以及酚类中的—OH基团拉伸振动;2 900 cm−1处的峰对应—CH2的伸缩振动;1 200~1 650 cm−1的多个谱带对应典型的C—N杂环伸缩振动。此外,对于CN600,801 cm−1处的峰对应C—S的环呼吸振动,而随着热解温度的升高,硫元素挥失,故其他CNx中未发现该结构。
图2(c)中的CNx的拉曼光谱显示出明显的D峰和G峰,且峰间距较大,表明CNx均为短程有序结构。D峰与G峰的强度比(ID/IG)通常用于分析碳材料的缺陷度。上述结果表明,随着制备温度的升高,CNx的ID/IG逐渐增大,也说明催化剂缺陷度增加。XPS表征结果表明, CN600和CN700中存在氮元素(图3(a))。而当制备温度高于800 ℃时,催化剂中无法检测到氮。N2脱附/吸附分析结果表明,除CN600为30.07 m2·g−1外,其余催化剂的比表面积均在85~95 m2·g−1。孔径分析结果表明,CNx的孔结构主要为介孔,且分布在30~40 nm。
-
在温度为(25±0.5) ℃、BPA浓度为40 μmol·L−1、CNx投加量为0.1 g·L−1、初始pH为6.5±0.1的条件下进行了BPA的吸附实验。如图3(c)所示,CN600、CN700、CN800、CN900、CN1000对BPA的去除率为3.4%、20.0%、27.42%、24.91%、27.2%,这表明所制备催化剂具有一定的BPA吸附性能。在上述实验条件下额外加入0.4 mmol·L−1的PMS进行BPA降解实验,结果见图3(d)。CN600、CN700、CN800、CN900、CN1000对BPA的总去除率分别提升至41.8%、96.6%、90.82%、75.25%和63.0%,这表明催化剂具有良好的PMS活化性能。
结合拉曼光谱和XPS精细谱分析,尽管CN600具有较高的氮掺杂量,但相同条件下其PMS活化性能却远低于氮含量较低的CN700。这表明催化剂中氮杂质缺陷的类型对催化性能有直接的影响。如图3(b)和表1所示,CN600中的氮杂质缺陷主要为吡啶氮(PDN),其含量远远高于吡咯氮(PLN)和石墨氮(GPN),而CN700中吡咯氮和石墨氮的含量占比更高。因此,可以推测,高含量的吡啶氮杂质缺陷不利于污染物去除,而吡咯氮和石墨氮杂质缺陷有较强的PMS活化性能。此外,CN700的缺陷度(ID/IG=0.975 0)小于CN600的缺陷度(ID/IG=0.981 6)。CN600中大量热力学不稳定的结构以及前驱体中—NH基团在高温状态下的易挥发性是导致缺陷度变化的可能原因(图3(e)与图3(f))。当热解温度大于(含)800 ℃时,CNx中的氮含量已无法检出。这进一步证明了含氮基团在高温下的不稳定性。值得注意的是,CN800(ID/IG=1.002 6)在没有氮掺杂的状态下仍然表现出了优异的催化性能。可以推测,随着热解温度的升高,氮杂质缺陷的损失会形成空穴或拓扑缺陷,这些缺陷同样具有良好的催化性能。CN900(ID/IG=1.011 6)表现出了最高的缺陷度,但其催化性能弱于CN800,这可能是由于高温下具有高催化活性的缺陷点位部分转化为具有低催化活性的点位,而CN1000(ID/IG=0.988 5)进一步降低的BPA降解性能证明了这一推论。因此,高温下的结构重组会导致缺陷度降低,进而影响催化剂的PMS活化性能,选择合适的制备温度对于提高催化剂性能尤其重要。
-
图4(a)反映了不同的催化剂投加量对BPA降解性能的影响。使用CN700为催化剂,在反应温度为(25±0.5) ℃、BPA浓度为40 μmol·L−1、PMS浓度为0.4 mmol·L−1、初始pH为6.5±0.1的条件下,催化剂的投加量对BPA的降解效率有显著的影响。当投加量为0.05 g·L−1时,BPA的去除率降低至53.2%;而当投加量增大至0.2 g·L−1时,BPA可在10 min内实现完全去除。这是因为,随着催化剂投加量的提高,可提供大量PMS活化所需要的活性点位,使PMS活化效率得到大幅度提升。这是BPA降解效率得到大幅度提高的主要原因[22-23]。
图4(b)反映了不同PMS浓度对BPA降解性能的影响。在反应温度为(25±0.5) ℃、BPA为40 μmol·L−1、CN700投加量为0.1 g·L−1、初始pH为6.5±0.1的条件下,当PMS投加量由0.2 mmol·L−1提升至0.8 mmol·L−1时,BPA的降解率由90.6%提升至100%,对应一级动力学常数由0.039 min−1提升为0.140 min−1。这表明较高PMS浓度可提供额外的传质推动力,能促进PMS在催化剂表面的吸附,实现对BPA更高效的降解。然而,当PMS浓度进一步提升至2.0 mmol·L−1时,其BPA降解效率以及一级动力学常数并没有得到对应的提升,反而轻微降低到0.136 min−1。这表明过量PMS的对污染物的降解有一定的抑制作用。该结果与文献报导的结果类似[24],过量PMS的解离平衡会形成大量SO5·-,SO5·-与活性氧化物种之间直接的反应是导致BPA降解受到抑制的主要原因。因此,适宜的PMS投加量选择尤为重要。
图4(c)反映了初始pH变化的影响。在反应温度为(25±0.5) ℃、BPA为40 μmol·L−1、PMS为0.4 mmol·L−1、CN700投加量为0.1 g·L−1时,初始pH的变化对BPA的降解有一定的影响。当初始pH分别为10.0、9.0、6.5、5.0和4.0时,对应的BPA降解率为90.55%、92.69%、97.09%、98.08%和100%。可以看出,BPA的降解率随着初始pH的升高有轻微的降低。这是由于随着初始pH的升高,溶液中OH−浓度随之升高。此外,PMS在水中以阴离子HSO5−形式存在,PMS的活化首先需要实现其在催化剂表面的有效吸附,而溶液中大量共存的OH−会通过竞争吸附占据催化剂表面的活性点位,导致PMS吸附受限,BPA的降解效率降低。而在酸性条件下,由于较高的H+活度,催化剂表面易发生质子化现象,从而通过静电作用力促进PMS的吸附,进而导致在较低初始pH下达到较好的BPA降解效果。
在反应温度为(25±0.5) ℃、BPA为40 μmol·L−1、PMS为0.4 mmol·L−1、CN700投加量为0.1 g·L−1、初始pH为6.5±0.1的反应条件下,分别考察了Cl−、
${\rm{NO}}_3^{-} $ 、${\rm{HPO}}_4^{2-} $ 、${\rm{HCO}}_3^{-} $ 等共存阴离子各自对BPA降解效果的影响(图4(d))。在Cl−、${\rm{NO}}_3^{-} $ 和$ {\rm{HPO}}_4^{2-}$ 等阴离子浓度为1.5 mmol·L−1的条件下,BPA的降解率有轻微的下降。这主要是由于:一方面,共存阴离子在催化剂表面的竞争吸附所导致;另一方面,Cl−与PMS反应会形成氧化性能较低的活性卤素(式(2)~式(4))。而有文献报道,${\rm{HPO}}_4^{2-} $ 的共存会对$ {\rm{SO}}_4^{-}\cdot $ 和·OH−等自由基产生掩蔽作用(式(5)和式(6)),导致BPA降解率的降低[25]。值得注意的是,$ {\rm{HCO}}_3^{-}$ 的共存会产生较为显著的抑制作用。除竞争吸附以外,${\rm{HCO}}_3^{-} $ 的共存可轻微提升溶液pH,造成BPA的降解效率下降。这与前述碱性条件下BPA降解效率下降的原因类似。 -
通过EPR分析和活性物种淬灭实验对CN700/PMS反应体系中的活性物种进行了鉴别。以5,5-二甲基-1-吡咯啉-N-氧化物(5,5-dimethyl-1-pyrroline N-oxide, DMPO)作为自旋捕获剂进行EPR分析,结果见图5(a)。在图5(a)中可观察到
${\rm{SO}}_4^{\cdot -} $ 和·OH的自由基信号,表明这些自由基可能参与了反应。甲醇(methanol)通常用于SO4·−和·OH的淬灭,而叔丁醇(tertiary-butyl alcohol, TBA)通常用于·OH的淬灭[29]。本研究分别使用浓度为200 mmol·L−1的甲醇与叔丁醇作为淬灭剂,考查其对BPA降解效果的影响。如图5(d)所示,溶液中的甲醇和叔丁醇均对BPA的降解有轻微的抑制作用。这表明上述2种自由基对BPA的降解有一定的贡献,但不是主导作用。通过2,2,6,6-四甲基哌啶(2,2,6,6-tetramethylpiperidine, TEMP)作为自旋捕获剂,可得到1O2的特征峰(图5(b))。随着反应的逐步进行,1O2特征峰的强度逐渐增大,表明催化剂在活化PMS时体系中产生了大量的1O2。使用浓度为8 mmol·L−1的L-组氨酸和NaN3作为1O2的淬灭剂[26],BPA的降解率大幅度降低至33.9%。这表明1O2对于BPA的降解具有重要的作用。同时,如图5(c)所示,使用DMPO作为自旋捕获剂,在甲醇作为溶剂的条件下可以检测到体系中的超氧自由基O2·−。而使用浓度为8 mmol·L−1的对苯醌(p-Bq)作为淬灭剂[27],发现BPA的去除率下降到18.0%,其降解反应受到严重的抑制。电化学开路电压(open-circuitpotential,OCP)的分析结果如图5(e)所示。催化剂界面的OCP随着体系中加入的PMS浓度的增加而升高,这表明催化剂可以有效地吸附PMS,形成了催化剂/PMS复合体,为后续的活化提供了先决条件。线性伏安曲线(linear sweep voltammetry,LSV)分析结果如图5(f)所示。在相同外电势的条件下,催化剂与吸附态PMS之间存在着电子传递过程。当BPA额外加入时,电流密度降低,表明BPA与催化剂/PMS复合体之间也存在着电子传递。该结果说明,催化剂/PMS复合体相比BPA具有更高的电位,可导致BPA分子结构中的电子向催化剂/PMS复合体传递,从而导致BPA的降解。这与恒电位电流-时间曲线分析的结果(图5(g))一致。
基于密度泛函理论(density functional theory,DFT),构建了常规碳结构、吡咯氮、吡啶氮、以及本征缺陷等5种不同缺陷结构的催化剂界面[28]。图6(a)中列出了上述界面各自对PMS吸附能(E吸附)。石墨氮结构具有优异的吸附PMS能力,其吸附能E吸附为-3.64 eV。此外,本征缺陷(E吸附=-1.77 eV)和吡咯氮缺陷点位(E吸附=-1.63 eV)也具有良好的PMS吸附能力。值得注意的是,吡啶氮缺陷点位对PMS的吸附能(E吸附=-1.28 eV)甚至低于常规碳结构(E吸附=-1.32 eV)。因此,可推测吡啶氮点位不是PMS的有效活化点位,这与前述XPS分析结果一致。该结果也进一步解释了高含氮量催化剂(CN600)具有较差PMS活化与BPA降解效果的原因。由图6(a)可以看出,在吡咯氮和本征缺陷点位附近的PMS中的S-O键分别被拉长了41.1%和28.9%。这表明这2种缺陷点位可能会促进S-O键的断裂,导致PMS的非对称裂解[29-30],生成O2·−。ATR-FTIR的结果同样证明了这一推测(图6(b)),图中1 100 cm−1和1 250 cm−1处峰分别对应了PMS中的S=O和S—O振动。当催化剂加入后,由于PMS与催化剂的结合,S—O的特征振动模式产生了改变并发生了从1 250 cm−1到1 195 cm−1红移,并伴随峰强的显著降低。这进一步证实了催化剂表面吸附态PMS的S—O断裂。
综上所述,CNx/PMS体系具体机理如下:首先,PMS被吸附到催化剂表面,形成催化剂与PMS的复合体。电子从催化剂向吸附态PMS的传递,导致PMS中S-O键断裂并生成O2·−(式(7))。形成的O2·−可进一步转化为1O2(式(8)),从而实现BPA的降解。该途径与对苯醌淬灭实验的结果一致,这解释了O2·−间接参与BPA降解过程的机制。同时,BPA被催化剂吸附后,由于催化剂/PMS复合体的高电位,BPA分子结构中的电子向复合体转移,可同样导致的BPA的降解。因此,CNx/PMS对BPA的高效降解主要通过1O2的高效生成和电子传递机制实现。
-
1)所制备的CNx与PMS的耦合可实现水中BPA的高效降解。随催化剂投加量和PMS投加量的增大,BPA的降解效率增加,但过量投加的PMS对BPA的额外降解无显著作用。
2)增加碳催化剂中的吡咯氮和石墨氮杂质缺陷活性点位,对于其PMS活化性能的提升有积极的作用。
3)碳催化剂中高活性的本征缺陷点位对于PMS的活化具有显著的促进作用。
4) CNx/PMS对BPA的高效降解主要通过高效生成的1O2和电子传递机制共同作用实现。
氮掺杂碳催化剂活化过一硫酸盐的活性位点分析及其对双酚A的降解机制
Analysis of active sites in nitrogen-doped carbocatalysts for peroxymonosulfate activation and the degradation mechanism of bisphenol A
-
摘要: 为了探究氮掺杂碳催化剂中不同类型缺陷点位在活化过一硫酸盐((PMS))时的反应活性,以碳黑和二氰二胺混合物为前驱体,通过热解得到了一系列不同氮掺杂量的碳催化剂(CNx),并对所制备催化剂的缺陷度、化学组分以及PMS活化性能进行了研究。结果表明,增加碳催化剂中的高活性氮杂质缺陷点位可有效促进催化剂的PMS活化性能;不同本征缺陷点位对PMS活化性能也表现出显著差异。活性物种淬灭实验、顺磁共振分析和电化学分析等结果表明,CNx/PMS体系对双酚A(BPA)的降解过程遵循以单线态氧(1O2)为主导的非自由基途径,催化剂表面的电子传递机制也有一定贡献。以上研究结果可为识别氮掺杂碳催化剂中的活性点位和高活性催化剂的定向合成提供参考。Abstract: In order to understand the reactivity of different defect sites in nitrogen-doped carbocatalysts with peroxymonosulfate (PMS), a series of carbocatalysts (CNx) with different nitrogen contents were prepared by pyrolyzing a mixture precursor consisted of carbon black and dicyandiamide. The defect degree, chemical composition and PMS activation performance of CNx were studied. The results showed that increased nitrogen defect sites with high-reactivity could promote the activation of PMS, and different intrinsic defect sites also exhibited significant differences in PMS activation. The results of reactive oxidation species quenching experiment, electron paramagnetic resonance analysis and electrochemical analysis showed that the bisphenol A (BPA) degradation by CNx / PMS was a non-radical pathway process, which was dominated by singlet oxygen (1O2), and the electron transfer mechanism also contributed to some extent. The above results could shed light on the identification of active sites in nitrogen-doped carbocatalysts and the oriented synthesis of catalysts with high catalytic activity.
-
Key words:
- AOPs /
- PMS /
- nitrogen-doped carbocatalysts /
- nitrogen defect /
- intrinsic defect
-
-

图 3 CNx的吸附、氧化性能、XPS分析和热重-红外联用分析
Figure 3. Performance of adsorption, catalytic PMS for BPA degradation by CNx/PMS system, XPS analysis and TG-FTIR analysis of CNx

图 4 CNx活化PMS降解BPA的影响因素分析
Figure 4. Effects of various reaction conditions on BPA degradation by CNx/PMS system
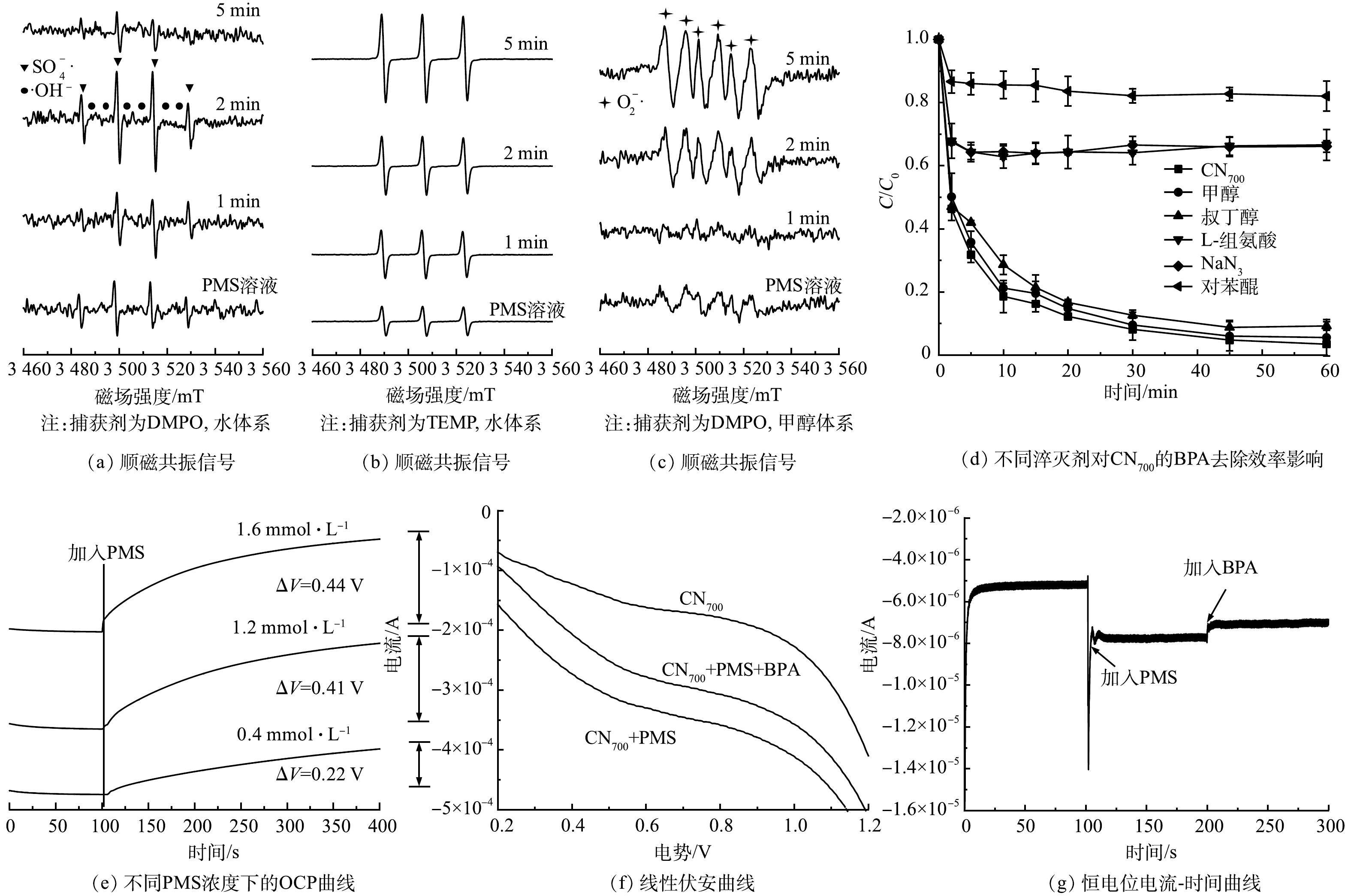
图 5 CNx的顺磁共振、淬灭实验和电化学分析
Figure 5. EPR, quenching experiment and electrochemical analysis of CNx
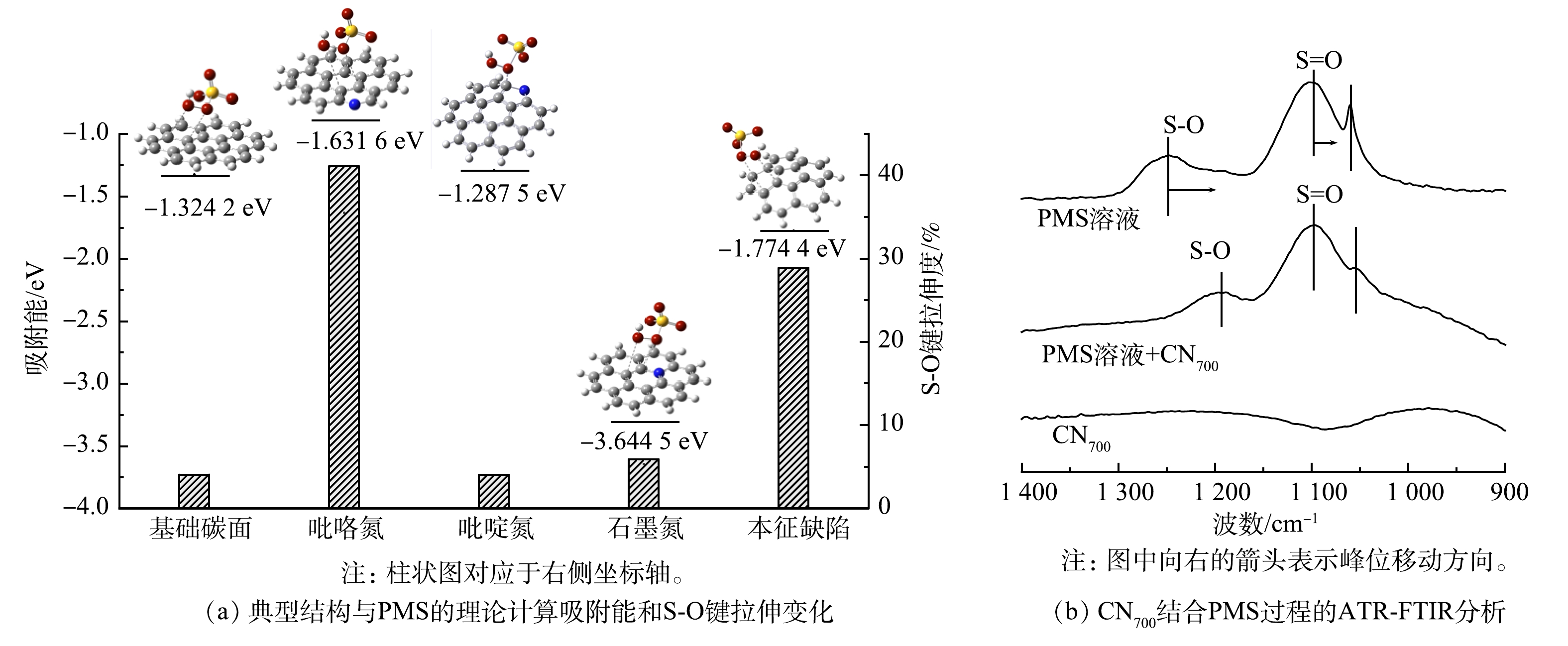
图 6 CNx的理论计算和ATR-FTIR分析
Figure 6. Theoretical calculation results and ATR-FTIR analysis of CNx
表 1 XPS分析中各元素含量以及N1s精细谱的各组分含量
Table 1. Content of each element in XPS analysis and the content of each component in N1s spectrum %
样品 C O N PLN PDN GPN Oxide-N CN600 86.94 3.44 9.63 2.92 5.96 0.54 0.21 CN700 96.02 2.35 1.64 0.31 0.79 0.15 0.39 CN800 97.35 2.65 - - - - - CN900 97.62 2.38 - - - - - CN1000 97.80 2.20 - - - - -  下载: 导出CSV
下载: 导出CSV
-
[1] 孟琪莉, 孙冲. 高级氧化技术在工业难降解有机废水处理中的应用研究进展[J]. 工业用水与废水, 2021, 52(3): 1-5. [2] 陆恬奕, 李宇, 徐瑞, 等. 高级氧化技术水处理研究进展[J]. 当代化工, 2021, 50(5): 1257-1260. [3] WANG J, WANG S. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 2018, 334: 1502-1517. doi: 10.1016/j.cej.2017.11.059 [4] 吴秀, 方迪, 危亚云, 等. 热活化过一硫酸盐调理强化厌氧消化污泥脱水的研究[J]. 环境科学学报, 2021, 41(11): 4547-4553. [5] 段书乐, 马婧捷, 党宁, 等. 紫外高级氧化工艺降解水溶液中的人工甜味剂[J]. 环境科学学报, 2020, 40(12): 4289-4296. [6] 王肖磊, 吴根华, 方国东, 等. 过渡金属活化过硫酸盐在环境修复领域的研究进展[J]. 生态与农村环境学报, 2021, 37(2): 145-154. [7] 田婷婷, 李朝阳, 王召东, 等. 过渡金属活化过硫酸盐降解有机废水技术研究进展[J]. 化工进展, 2021, 40(6): 3480-3488. [8] DING Y, WANG X, FU L, et al. Nonradicals induced degradation of organic pollutants by peroxydisulfate (PDS) and peroxymonosulfate (PMS): Recent advances and perspective[J]. Science of the Total Environment, 2021, 765: 142794. doi: 10.1016/j.scitotenv.2020.142794 [9] ZHAO C, SHAO B, YAN M, et al. Activation of peroxymonosulfate by biochar-based catalysts and applications in the degradation of organic contaminants: A review[J]. Chemical Engineering Journal, 2021, 416: 128829. doi: 10.1016/j.cej.2021.128829 [10] CAI Q Q, LEE B C Y, ONG S L, et al. Fluidized-bed Fenton technologies for recalcitrant industrial wastewater treatment:Recent advances, challenges and perspective[J]. Water Research, 2021, 190: 116692. doi: 10.1016/j.watres.2020.116692 [11] ZHANG W, LI Y, FAN X, et al. Synergy of nitrogen doping and structural defects on hierarchically porous carbons toward catalytic oxidation via a non-radical pathway[J]. Carbon, 2019, 155: 268-278. doi: 10.1016/j.carbon.2019.08.071 [12] ENOKI T, FUJII S, TAKAI K. Zigzag and armchair edges in graphene[J]. Carbon, 2012, 50(9): 3141-3145. doi: 10.1016/j.carbon.2011.10.004 [13] WOHNER N, LAM P K, SATTLER K. Systematic energetics study of graphene nanoflakes: From armchair and zigzag to rough edges with pronounced protrusions and overcrowded bays[J]. Carbon, 2015, 82: 523-537. doi: 10.1016/j.carbon.2014.11.004 [14] MIELKE S L, TROYA D, ZHANG S, et al. The role of vacancy defects and holes in the fracture of carbon nanotubes[J]. Chemical Physics Letters, 2004, 390(4): 413-420. [15] LIU J, LIANG T, TU R, et al. Redistribution of π and σ electrons in boron-doped graphene from DFT investigation[J]. Applied Surface Science, 2019, 481: 344-352. doi: 10.1016/j.apsusc.2019.03.109 [16] ADIL S, KIM W S, KIM T H, et al. Defective, oxygen-functionalized multi-walled carbon nanotubes as an efficient peroxymonosulfate activator for degradation of organic pollutants[J]. Journal of Hazardous Materials, 2020, 396: 122757. doi: 10.1016/j.jhazmat.2020.122757 [17] WANG J, DUAN X, GAO J, et al. Roles of structure defect, oxygen groups and heteroatom doping on carbon in nonradical oxidation of water contaminants[J]. Water Research, 2020, 185: 116244. doi: 10.1016/j.watres.2020.116244 [18] OUYANG D, CHEN Y, YAN J, et al. Activation mechanism of peroxymonosulfate by biochar for catalytic degradation of 1, 4-dioxane: Important role of biochar defect structures[J]. Chemical Engineering Journal, 2019, 370: 614-624. doi: 10.1016/j.cej.2019.03.235 [19] ZHANG H, LI X, ZHANG D, et al. Comprehensive electronic structure characterization of pristine and nitrogen/phosphorus doped carbon nanocages[J]. Carbon, 2016, 103: 480-487. doi: 10.1016/j.carbon.2016.03.042 [20] KANG B, SHI H, WANG F-F, et al. Importance of doping site of B, N, and O in tuning electronic structure of graphynes[J]. Carbon, 2016, 105: 156-162. doi: 10.1016/j.carbon.2016.04.032 [21] ZHOU X, ZHAO C, WU G, et al. DFT study on the electronic structure and optical properties of N, Al, and N-Al doped graphene[J]. Applied Surface Science, 2018, 459: 354-362. doi: 10.1016/j.apsusc.2018.08.015 [22] HU P, SU H, CHEN Z, et al. Selective Degradation of organic pollutants using an efficient metal-free catalyst derived from carbonized polypyrrole via peroxymonosulfate activation[J]. Environmental Science & Technology, 2017, 51(19): 11288-11296. [23] QI F, CHU W, XU B. Ozonation of phenacetin in associated with a magnetic catalyst CuFe2O4: The reaction and transformation[J]. Chemical Engineering Journal, 2015, 262: 552-562. doi: 10.1016/j.cej.2014.09.068 [24] DU J, BAO J, LIU Y, et al. Efficient activation of peroxymonosulfate by magnetic Mn-MGO for degradation of bisphenol A[J]. Journal of Hazardous Materials, 2016, 320: 150-159. doi: 10.1016/j.jhazmat.2016.08.021 [25] ZHANG T, CHEN Y, WANG Y, et al. Efficient peroxydisulfate activation process not relying on sulfate radical generation for water pollutant degradation[J]. Environmental Science & Technology, 2014, 48(10): 5868-5875. [26] LIU X, CHEN Y, YAO Y, et al. Iodine-doped carbon fibers as an efficient metal-free catalyst to activate peroxymonosulfate for the removal of organic pollutants[J]. Catalysis Science & Technology, 2018, 8(21): 5482-5489. [27] SUN P, LIU H, FENG M, et al. Nitrogen-sulfur co-doped industrial graphene as an efficient peroxymonosulfate activator: Singlet oxygen-dominated catalytic degradation of organic contaminants[J]. Applied Catalysis B:Environmental, 2019, 251: 335-345. doi: 10.1016/j.apcatb.2019.03.085 [28] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 16 Rev. C. 01 [M]. Wallingford, CT. 2016. [29] LI X, YANG S, DZAKPASU M, et al. Galvanic corrosion of zero-valent iron to intensify Fe2+ generation for peroxymonosulfate activation[J]. Chemical Engineering Journal, 2021, 417: 128023. doi: 10.1016/j.cej.2020.128023 [30] WANG G, NIE X, JI X, et al. Enhanced heterogeneous activation of peroxymonosulfate by Co and N codoped porous carbon for degradation of organic pollutants: The synergism between Co and N[J]. Environmental Science:Nano, 2019, 6(2): 399-410. doi: 10.1039/C8EN01231H -
 点击查看大图
点击查看大图

计量
- 文章访问数: 7238
- HTML全文浏览数: 7238
- PDF下载数: 149
- 施引文献: 0










