

 下载:
下载:
-
太阳能制氢(H2转换)被认为是解决环境问题和全球能源危机的一种有吸引力和可持续的技术. 通过用清洁可持续的氢能代替化石燃料来缓解环境污染和能源问题[1 − 3]. 但是目前光解水制氢的催化剂还存在诸多问题导致远没有达到工业应用的门槛. 例如,催化剂的长期稳定性不足,反应仍然依赖贵金属助催化剂,牺牲剂(如三乙醇胺[4]、Na2S/Na2SO3[5 − 6]或乳酸[7]),催化剂活性降低难以重复利用等问题无疑提高了光解水制氢催化剂的使用成本.
硫化镉(CdS)及其杂化物被用于光催化水分解以生产清洁的H2燃料. 由于其非常合适的带隙宽度和价带位置与其他金属硫化物相比表现出最佳的制氢结果[8]. 然而,由于反应位点不足和光生电子和空穴的快速复合,原始CdS上的产氢活性依然难以令人满意. 为了克服这些障碍,负载助催化剂已被证明是最有前途的方法之一,它不仅可以为光催化氧化还原反应提供额外的活性位点,还可以形成异质结以提高电荷分离效率[9]. 贵金属Pt被认为是目前最有效的助催化剂,但它昂贵的价格限制了它的实际应用. 因此,迫切需要开发其他无贵金属材料与CdS偶联用于光催化产氢来提升成本效益. Liu等[10]在CdS上负载黑磷量子点(BPQDs)做助催化剂,使用简单的超声方法制备了BPQDs/CdS二元复合材料,其在可见光下产氢效果达到9.9 mmol·g−1·h−1,是所用BPQDs和CdS的99倍和5.5倍. 表明了BP作为助催化剂可以显著提升CdS材料光催化产氢性能,但是制备BPQDs的过程十分繁琐,并且产率极低,不利于工业化生产.
本文提出了新开发的简单溶剂热法制备黑磷红磷(BP/RP)-CdS异质结光催化剂被应用于改善CdS的产氢活性. BP/RP和CdS构成异质结,既能提高黑磷的稳定性,还优化了复合材料的电子转移. 合成的BP/RP -CdS异质结光催化剂在光催化水分解的应用中,最高产率为10.3 mmol·g−1·h−1,产氢速率是纯CdS材料的2.21倍,超过了Pt-CdS的10.1 mmol·g−1·h−1产氢产率. 本研究表明,无贵金属材料的BP/RP异质结可以取代贵金属Pt作为CdS的助催化剂,降低的催化剂成本为BP/RP-CdS的工业化生产应用提供了可能性.
-
红磷、乳酸(90%)购于天津大茂化学试剂厂,氯化镉半五水合物、硫脲、二乙烯三胺均购于上海阿拉丁生化科技股份有限公司,无水乙醇购于四川西陇科学股份有限公司. 除注明含量的试剂外,均为分析纯,实验所用去离子水为实验室自制.
扫描电子显微镜(SEM,ZEISS Gemini 300)、透射电子显微镜(TEM,Talos F200S)用于样品形貌表征;X射线衍射仪(XRD,日本理学Ultima IV )、拉曼光谱仪(Raman,Renishaw)、X射线光电子能谱(XPS,Thermo Fisher Escalab 250Xi)对样品结构进行了表征;使用UV 3600I plus测试了样品的紫外可见漫反射光谱(UV-Vis DRS);光电化学(PEC)测量是在电化学工作站CHI760E(上海辰华仪器公司)中进行的,使用的350 W氙灯(北京镁瑞臣科技有限公司HSX-F300)用于模拟太阳光. 涂附有催化剂样品的导电玻璃直接用作光阳极,铂片用作对电极,即光电阴极,而Ag/AgCl电极用作参比电极.
-
使用商业红磷作为原料,3 g商业红磷预先置于水热反应釜中,加入60 mL去离子水,在200 ℃条件下保温12 h,离心去除上清液,置于真空干燥箱中,60 ℃下真空干燥8 h,得到了去除氧化层的红磷记为RP,用作后续制备原料.
-
称取600 mg RP转移至水热反应釜中,加入60 mL乙烯三胺作为反应溶剂,在100—160 ℃条件下保温9—15 h,将反应产物用去离子水和无水乙醇分别洗涤3次以去除反应溶剂,将离心得到的产物放入真空干燥箱中,60 ℃条件下干燥24 h,得到BP/RP样品.
-
称取3.29 g CdCl2·2.5H2O溶解在60 mL二乙烯三胺中搅拌20 min,然后称取3.45 g硫脲分3份缓慢倒入二乙烯三胺溶剂中. 继续搅拌20 min,随后取出转子,反应在100 mL水热反应容器中进行,将反应器放入烘箱中,在 160 ℃下保温12 h. 冷却后使用超纯水和无水乙醇交替离心洗涤3次,之后将样品放入真空干燥箱中60 ℃下干燥12 h,得到CdS样品.
-
BP/RP-CdS材料的制备与CdS的制备基本一致. 采用二乙烯三胺为溶剂,按比例加入RP与CdCl2·2.5H2O于100 mL水热反应釜中,共同搅拌20 min,然后分3份缓慢加入硫脲. 继续搅拌20 min后将反应器放入烘箱中,在 160 ℃下保温12 h,冷却后使用超纯水和无水乙醇交替离心洗涤3次,之后将样品放入真空干燥箱中60 ℃下干燥12 h,得到BP/RP-CdS样品.
-
在带有石英盖的柱状石英反应釜中,加入10 mg光催化剂、5 mL 10%(体积分数)乳酸(作为牺牲电子供体)和45 mL去离子水,将配置好的水溶液反应体系置于反应釜中超声5—10 min 使催化剂分散均匀,随后将反应釜接在反应装置的气路上,将整个反应装置抽真空后进行光催化性能测试. 在光催化系统中,使用带有AM1.5滤光片的300 W氙灯光照系统提供光源,用来模拟太阳光. 光源的初始电流强度设置为16 A,为防止溶液汽化进入色谱,同时排除温度对实验的影响,采用了278 K的冷凝循环水为光催化系统保温. 进行测试时,在连接石英封闭真空循环系统的石英反应池中进行,所产生的气体每间隔0.5 h采一次样送入气相色谱仪中自动采集和分析. 每组反应时间为5 h.
-
图1a是CdS复合材料的SEM图像,可以看出溶剂热制得的CdS呈团簇向外放射生长出了许多宽约20—50 nm长约几百纳米的棒状纳米结构. 在BP/RP-CdS复合材料的SEM图中出现比CdS的更加粗大的纳米棒(图1b),这可能是因为加入的BP/RP纳米片附着在CdS表面形成的纳米棒尺寸增大. 对图1c的BP/RP-CdS复合样品进行了EDS能谱面扫,得到的各元素的分布情况如图1d所示,可以看出Cd,S和P元素均匀的分布在CdS表面,说明了BP/RP成功复合到了CdS表面,同时还检测到了轻微的O元素,说明复合材料有一定的氧化.
CdS(图2a)和BP/RP-CdS复合材料(图2b)的TEM图像显示出生长方向基本一致纳米棒团簇. 对复合材料的表面进行HRTEM观察,图2e显示CdS条纹间距为0.353 nm,这与六方相CdS的(100)晶面间距符合[11]. 在CdS晶体表面观察到更细小的晶格条纹,条纹间距为0.198 nm和0.213 nm,分别与BP的(022)和(002)晶面间距相匹配[12]. TEM图像显示清晰的硫化镉和黑磷晶格条纹说明了溶剂热法得到了高结晶度的BP/RP-CdS复合材料. 对图2f的EDS能谱面扫结果同样可以看出P在CdS基底上均匀分布.
由图3可以看出,溶剂热法制得的CdS特征峰符合六方相硫化镉晶型(PDF # 41-
1049 ),分别对应心面立方CdS的(100)、(002)、(101)、(102)、(110)、(103)和(112)平面反射[13]. 在XRD图中并没有BP特征峰显示是由于复合材料中BP/RP的添加量较小. 在XRD图中可以观察到CdS材料的最强峰(002)在26.54°,而3%、5%、10%BP/RP-CdS的衍射峰分别为26.48°、26.50°和26.52°,均出现了略微的左移. 这是因为P的原子半径大于S,所以当CdS纳米棒与BP/RP耦合时,XRD衍射峰向左移动[14].在对CdS和BP/RP-CdS复合材料进行了XPS测试,如图4a所示. Cd、S、P、C的特征峰在BP/RP-CdS复合材料的全谱上都可以检测到. 图4b是BP/RP和复合材料的P 2p谱,由于复合材料中P含量较少,所以XPS信号比较弱,这与XRD结果匹配. 对比BP/RP材料,复合材料的P 2p峰向高结合能有2 eV的位移,这说明P元素在复合后电子密度下降. 在复合材料中Cd 3d的高分辨率XPS光谱表现出两个峰,结合能值分别为403.8 eV和410.6 eV,这是Cd 3d5/2和Cd 3d3/2特有的双峰结构[15]. 两个峰之间的自旋轨道耦合为6.8 eV,证实了异质结光催化剂中的Cd主要以Cd2+形式存在. 对比纯的CdS,双峰出现了0.3 eV的偏移,这与P 2p轨道的偏移方向相反,这说明了BP/RP-CdS复合材料中电子向CdS的转移. 在CdS材料和BP/RP-CdS复合材料的S 2p高分辨率XPS谱中,BP/RP-CdS复合材料出现结合能值为160.2 eV和161.5 eV的两个峰,分别分配给S 2p3/2、S 2p1/2[16]. 与CdS材料中S 2p3/2、S 2p1/2的结合能值相比有0.3 eV的偏移,也说明了在BP/RP-CdS复合材料中,电子更趋向于向CdS的转移.
-
光催化产氢实验是在仅添加乳酸牺牲剂的条件下测量. 无特别说明,光催化产氢实验均使用10 mg催化剂进行实验,并利用乳酸在CdS材料发生光腐蚀之前就能快速消耗光生空穴,从而确保有效的光生电子空穴的分离.
如图5a所示,探究了CdS、BP/RP-CdS、BP-CdS和RP-CdS的在5 h光催化中的产氢效率. 纯CdS的产氢效率只有0.047 mmol·h−1,BP/RP-CdS有最好的产氢效率(0.103 mmol·h−1),BP-CdS和RP-CdS的产氢效率差距不大,分别为0.072 mmol·h−1和0.074 mmol·h−1. 之后本文探究了不同BP/RP负载量对复合材料的产氢效率影响,如图5b所示,纯CdS在反应5 h时产氢量只有
0.0813 mmol,Pt作为助催化剂的Pt-CdS产氢量达到0.215 mmol,值得注意的是,CdS在0.5 h时出现产量下降的情况,这是由于硫化物表面堆积过多的光生空穴造成光腐蚀现象,使得光催化剂不稳定[17]. 在复合样品中,BP/RP的加入,提高了复合材料整体的光催化活性,各个比例的复合材料B/RP-CdS的产氢量均高于纯CdS,且复合材料中并没有出现光腐蚀造成的产量下降情况. 如图5c所示,在加入5%BP/RP时,产氢速率达到最高,为0.103 mmol·h−1,产氢速率是纯CdS材料的2.21倍,可以看出BP/RP的加入极大的提高了CdS的光催化产氢性能,超过了加入Pt助催化的Pt-CdS产氢速率为0.101 mmol·h−1. 对添加了BP/RP得到复合材料光催化产氢性能得到提高的可能原因包括表面性质的变化和异质结的形成[18 − 19]. 向CdS中添加BP/RP可以使电子更容易转移到表面并随后进行H2释放反应. 但是在加入10%BP/RP时,相比加入5%BP/RP的产氢效率出现了下降,可能是BP/RP的过量添加过度遮盖了CdS的表面影响了CdS的光吸收效果. 图5d是5%BP/RP-CdS复合材料在单次光催化产氢前后的XRD图. 在反应后的XRD峰值强度略微下降了,但是特征峰的位置没有出现变化,说明在光催化产氢实验中没有改变催化剂的结构. 在相同的实验条件下做了产氢的循环实验(图5e). 在3次产氢循环实验后,5%BP/RP-CdS与CdS分别表现出其初始PHE活性的95.4%、91.7%,通过以上结果可以看出溶剂热法制备的BP/RP-CdS材料表现出较好的光催化稳定性.用特定的光波长滤波镜片(400、450、500、550、600 nm)来测量BP/RP-CdS的表观量子产率(AQY). 使用以下公式计算:
式中,ν为反应速率(mol·s−1),K为反应转移电子数,在产氢反应中K=2,I为光功率密度(W·cm−2),A为入射光照面积(m2),λ为入射光波长(nm). 经过计算将AQY结果绘制到图5f中. 从图中可以看到,在400 nm滤光片下表现出11.4%的AQY,但在450 nm后迅速下降为2.12%,到600 nm时AQY降至1.20%.
-
紫外可见漫反射光谱(图6a)展示了BP/RP、CdS、BP/RP-CdS的最大吸收波长分别为792、567、591 nm. 可以看出BP/RP有极强的红外尾部吸收,CdS的吸收边最窄. BP/RP-CdS的吸收边相比CdS红移,说明BP/RP的加入使BP/RP-CdS复合材料的光吸收范围变大,并且在550 nm之后有明显的红外尾部吸收,表明复合材料具有更宽的吸收范围,具有更强的光响应能力.
图6b是Tauc plot法[20]转换固体紫外数据得到的CdS、BP/RP和BP/RP-CdS材料的带隙(Eg),分别为2.31、1.50、2.25 eV. 复合材料的Eg相比CdS略有减小.
图6c是380 nm激发波长光照射下CdS和BP/RP-CdS材料的PL光谱. 可以看出CdS在450 nm与750 nm处具有极强的光响应,BP/RP-CdS复合材料的光致发光强度低于CdS,说明BP/RP的加入显著抑制了光生电子和空穴的复合.
CdS和BP/RP-CdS材料在辐照时均表现出快速的光电流响应(图6d). BP/RP-CdS复合材料表现出比CdS更高的光电流响应. 这表明在CdS材料中添加BP/RP会增强光响应能力,激发更多的光生电子和空穴,从而提高BP/RP-CdS复合材料光催化产氢性能.
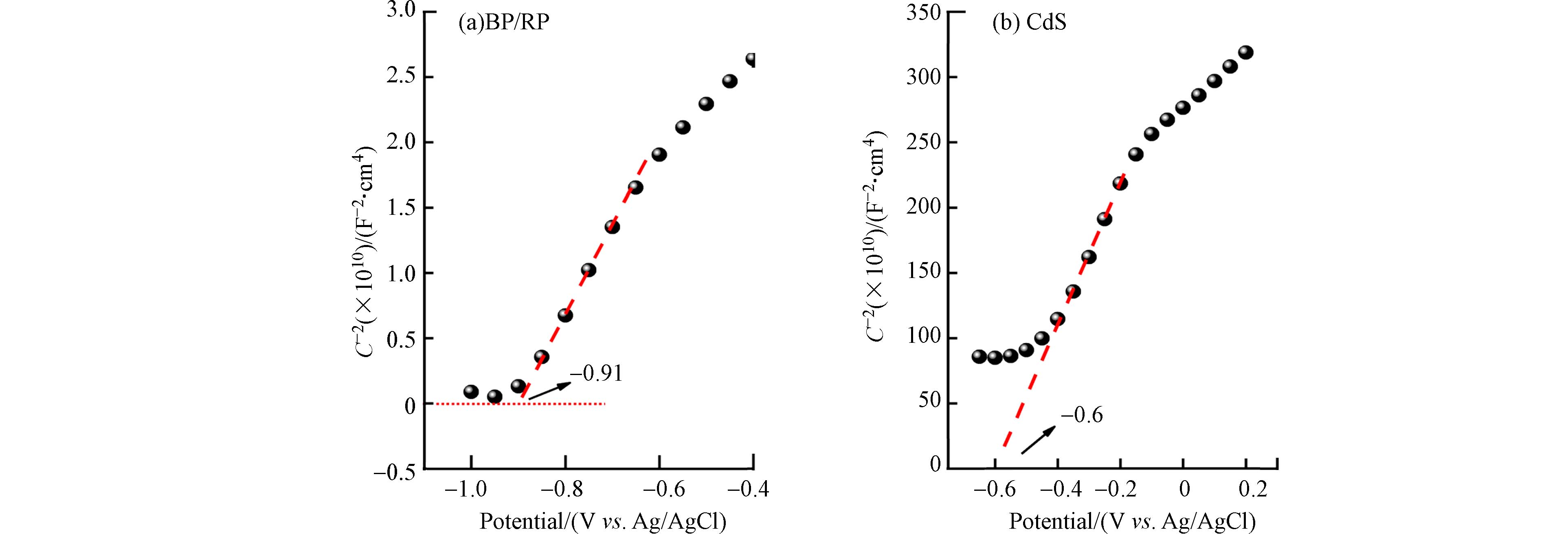
通过测出的莫特肖特基曲线(图7)得到了BP/RP与CdS的平带电位(Vf),分别为−0.91 eV和−0.60 eV. BP/RP与CdS的莫特肖特基曲线斜率都为正,证明它们都为n型半导体. 对于n型半导体来说,它的导带底EC比平带电位Vf要更负,EC=Vf -0.1 eV[21]. BP/RP与CdS材料在Ag/AgCl电极下的EC,分别为−0.70 eV和−1.01 eV. 将它们的EC换算成标准氢电极[22]. 将在Ag/AgCl参比电极测试得到的平带电位通过Nernst方程转化为可逆氢电极(RHE),Ag/AgCl参比电极转化为可逆氢电极(RHE):
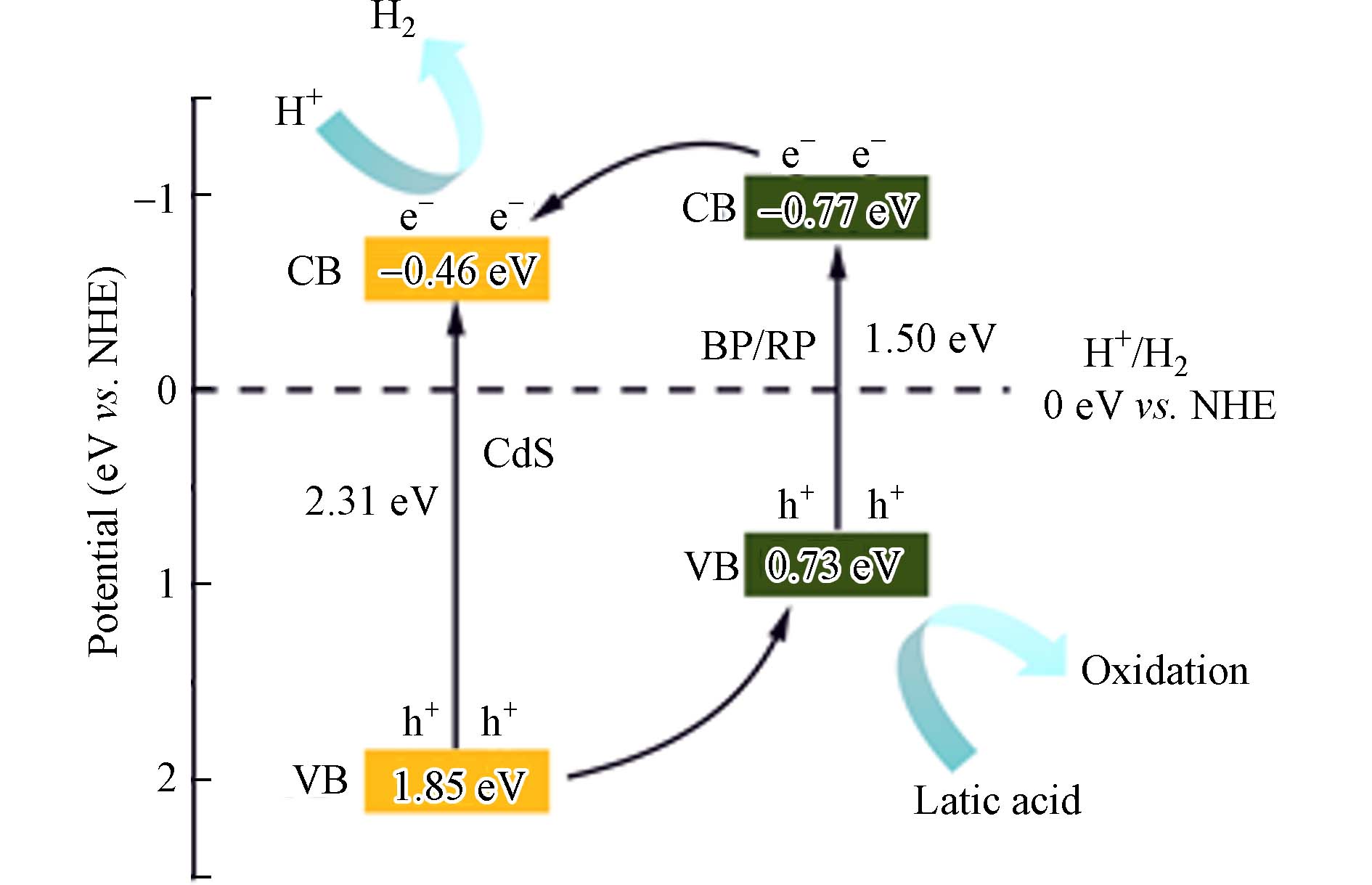
经过换算的BP/RP与CdS材料的导带电位分别为 −0.46 eV和−0.77eV在将材料的平带电位转化为导带电位,并结合固体紫外漫反射结果,可以得到半导体的能带图谱. 在紫外可见漫反射光谱中得到了BP/RP与CdS材料的Eg分别为1.50 eV和2.31 eV. 然后根据公式EV=EC+Eg可以求出对应材料的价带电位EV分别为0.73 eV和1.85 eV.
根据以上数据,确定了BP/RP-CdS复合材料能带结构,结合之前XPS部分的电子转移分析,可以得出BP/RP和CdS异质结材料的光催化机理如图8所示. 在反应体系中,乳酸作为牺牲剂,与光催化剂表面的空穴复合,发生氧化反应,在异质结材料的导带上光生电子作用下析出氢气. 在可见光照射下,CdS和BP/RP价带上的电子被激发并迁移到各自的导带. BP/RP导带上的光生电子迁移到CdS的价带并催化氢气的生成. 在CdS的价带上的空穴会转移到BP/RP材料的价带上,然后与乳酸结合之后被消除. 形成的Ⅱ型异质结中,电子向更正的导带电位转移,同时空穴转移到价带能级更高的半导体,这样的异质结构有效减少了电子-空穴对的接触和复合,从而有利于提高光催化产氢性能[23].
-
本文通过简便的一步溶剂热法成功制备BP/RP-CdSⅡ型异质结材料,证实了BP/RP的形成,并说明了BP/RP与CdS的紧密界面结合方式. 光催化产氢测试结果表明,加入5%BP/RP-CdS的材料产氢速率最高,达到了10.3 mmol·g−1·h−1,是CdS材料的2.21倍. 同一实验条件下,加入BP/RP助催化剂在效果上可以媲美贵金属Pt助催化剂. 异质结材料在3次产氢循环实验中仅有轻微下降,表现出了较好的稳定性. BP/RP的加入提高了整体光催化产氢的性能. 利用BP/RP异质结替代Pt贵金属作为助催化剂大大降低催化剂成本,为复合光催化剂的选择提供新方向.
一步溶剂热法制备BP/RP-CdS异质结材料的光催化产氢
One-step solvothermal preparation and photocatalytic performance of BP/RP-CdS heterojunction in hydrogen evolution
-
摘要: 本研究使用红磷(RP)、氯化镉、硫脲为反应原料,二乙烯三胺为反应溶剂,通过简单的一步溶剂热法使RP原位生长在硫化镉(CdS)上,同时部分转化为黑磷(BP),一步得到了BP/RP-CdS三元复合材料. 使用、扫描电子显微镜(SEM)、透射电子显微镜(TEM)、X射线光电子能谱(XPS),证明BP/RP-CdS材料形成了紧密接触结构,通过紫外漫反射及平带电位的结果计算出材料的带隙与价带导带位置,结合XPS中电子的转移说明形成了Ⅱ型异质结. 在乳酸水溶液中进行了光催化产氢评价. 结果表明,三元材料产氢速率最大为10.3 mmol·g−1·h−1,是CdS材料的2.21倍,与加Pt助催化剂的CdS材料产氢速率相当. BP/RP与CdS的紧密结合提高了光催化产氢活性.Abstract: In this study, BP/RP-CdS heterojunction materials were synthesized via a simple one-step solvothermal method employing red phosphorus, cadmium chloride and thiourea as reactants, with diethylenetriamine serving as the reaction solvent. During the process, red phosphorus underwent in-situ growth on CdS, with a portion of it was transforming into black phosphorus. Consenquently, a BP/RP-CdS ternary composite was obtained in a single step. Characterization techniques including transmission electron microscope, scanning electron microscope, and X-ray diffraction confirmed the close contact structure of the BP/RP-CdS material. The band gap and conduction band position were determined through UV diffuse reflection and flat band potential measurements. Moreover, XPS analysis indicated the formation of a type Ⅱ heterojunction. The photocatalytic hydrogen evolution performance was assessed in a lactic acid aqueous solution, revealingthat the ternary material exhibited a maximum hydrogen evolution rate of 10.3 mmol·g−1·h−1, surpassing that of CdS alone by 2.21 times and comparable to CdS with Pt co-catalyst. The integration integrationof BP/RP and CdS notably enhancesthe photocatalytic hydrogen evolution activity.
-
-

图 1 (a)CdS及(b)BP/RP-CdS复合材料的SEM图像,(c)BP/RP-CdS复合材料的EDS能谱扫描区域,及(d)各元素分布示意图(Cd、S、P、O)
Figure 1. SEM images of (a) CdS and (b) BP/RP-CdS composites, (c) EDS spectral scanning region of BP/RP-CdS composites and (d) distribution of each element (Cd、S、P、O)

图 2 CdS(a、b)及BP/RP-CdS(c—e)复合材料的TEM图像;(f—i)BP/RP-CdS复合材料的能谱面扫图
Figure 2. TEM image of CdS (a, b) and BP/RP-CdS (c—e) composites;(f—i) energy spectrum scan of BP/RP-CdS composites
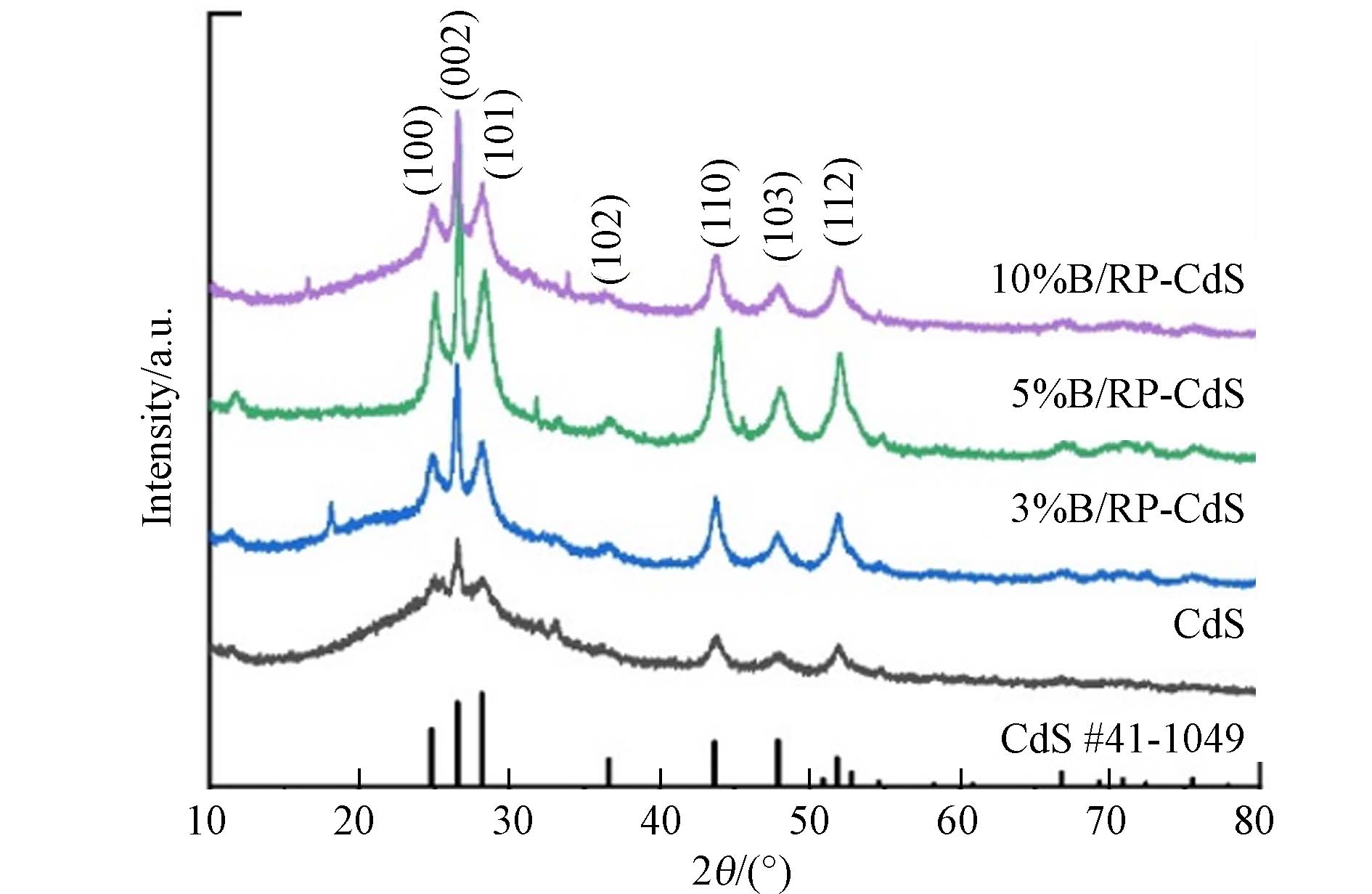
图 3 CdS及添加3%、5%、10% BP/RP的复合样品的XRD图像
Figure 3. XRD image of CdS and composite sample with 3%, 5%, 10% BP/RP

图 4 CdS和BP/RP-CdS复合材料的XPS能谱图
Figure 4. XPS energy spectrum of CdS and BP/RP-CdS composites

图 5 (a)CdS催化剂及磷复合CdS材料光催化产氢图;(b,c)CdS催化剂及BP/RP-CdS复合材料光催化产氢图;(d)5% BP/RP-CdS复合材料光催化产氢前后的XRD;(e)5% BP/RP-CdS与CdS样品的循环产氢图;(f)BP/RP-CdS的表观量子产率图
Figure 5. (a) Photocatalytic hydrogen evolution diagram of CdS catalyst and phosphorus composite CdS material; (b, c) photocatalytic hydrogen evolution diagram of CdS catalyst and BP/RP CdS composite material; (d) XRD before and after photocatalytic hydrogen evolution of 5% BP/RP CdS composite material; (e) cyclic hydrogen evolution diagram of 5% BP/RP CdS and CdS sample; (f) apparent quantum yield diagram of BP/RP CdS
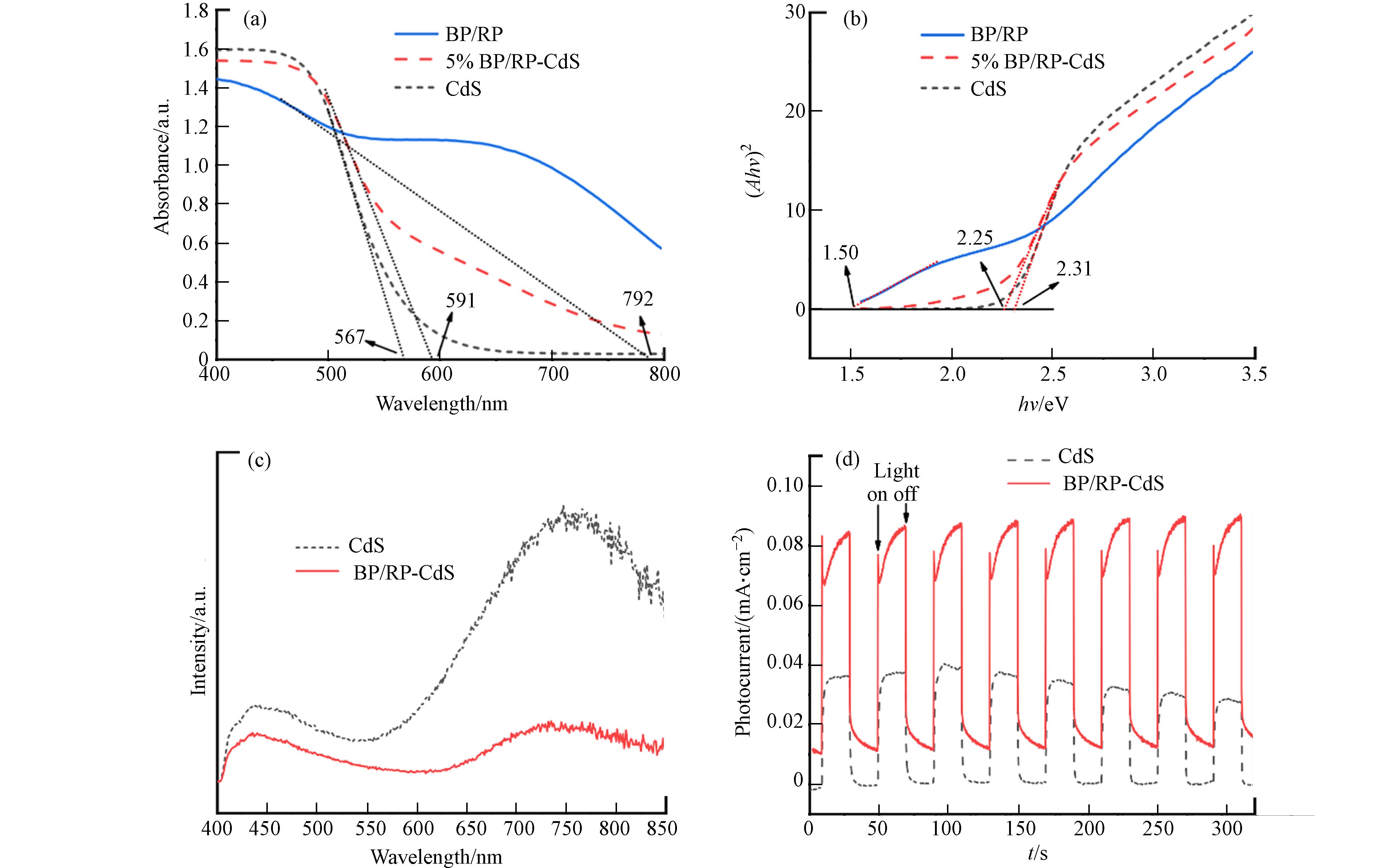
图 6 (a)BP/RP、CdS和BP/RP-CdS的紫外可见漫反射光谱、(b)BP/RP、CdS和BP/RP-CdS的Eg(c)CdS和BP/RP-CdS的稳态光致发光光谱(PL)(d)CdS和BP/RP-CdS在+0.1 V偏置电位下的光电流密度
Figure 6. (a) UV-visible diffuse reflectance spectra of BP/RP, CdS and BP/ RP-cds, and (b) band gaps of BP/RP, CdS and BP/ RP-CdS, (c) Steady-state photoluminescence spectroscopy (PL) of CdS and BP/RP-CdS, (d) Photocurrent density of CdS and BP/RP-CdS at +0.1 V bias potential
-
[1] BOUMERIAME H Da SILVA E S CHEREVAN A S et al. Layered double hydroxide (LDH)-based materials: A mini-review on strategies to improve the performance for photocatalytic water splitting[J]. Journal of Energy Chemistry, 2022, 64(1): 406-431 (in Chinese). [2] ZHANG K L, HU H J, SHI L T, et al. Strategies for optimizing the photocatalytic water-splitting performance of metal–organic framework-based materials[J]. Small Science, 2021, 1(12): 2100060. doi: 10.1002/smsc.202100060 [3] ABDUL NASIR J, MUNIR A, AHMAD N, et al. Photocatalytic Z-scheme overall water splitting: Recent advances in theory and experiments[J]. Advanced Materials, 2021, 33(52): e2105195. doi: 10.1002/adma.202105195 [4] ZHANG G P, CHEN D Y, LI N J, et al. Construction of hierarchical hollow Co9S8/ZnIn2S4 tubular heterostructures for highly efficient solar energy conversion and environmental remediation[J]. Angewandte Chemie, 2020, 132(21): 8332-8338. doi: 10.1002/ange.202000503 [5] HAO X Q, XIANG D Z, JIN Z L. Zn‐vacancy engineered S‐scheme ZnCdS/ZnS photocatalyst for highly efficient photocatalytic H2 evolution[J]. ChemCatChem, 2021, 13(22): 4738-4750. doi: 10.1002/cctc.202100994 [6] LIU Y Y, XIANG Z H. Fully conjugated covalent organic polymer with carbon-encapsulated Ni2P for highly sustained photocatalytic H2 production from seawater[J]. ACS Applied Materials & Interfaces, 2019, 11(44): 41313-41320. [7] LIU Y, NIU H T, GU W, et al. In-situ construction of hierarchical CdS/MoS2 microboxes for enhanced visible-light photocatalytic H2 production[J]. Chemical Engineering Journal, 2018, 339: 117-124. doi: 10.1016/j.cej.2018.01.124 [8] CHANDRASEKARAN S, YAO L, DENG L B, et al. Recent advances in metal sulfides: From controlled fabrication to electrocatalytic, photocatalytic and photoelectrochemical water splitting and beyond[J]. Chemical Society Reviews, 2019, 48(15): 4178-4280. doi: 10.1039/C8CS00664D [9] CAO S, CHEN Y, WANG C J, et al. Spectacular photocatalytic hydrogen evolution using metal-phosphide/CdS hybrid catalysts under sunlight irradiation[J]. Chemical Communications, 2015, 51(41): 8708-8711. doi: 10.1039/C5CC01799H [10] LIU F L, WANG Z, WENG Y X, et al. Black phosphorus quantum dots modified CdS nanowires with efficient charge separation for enhanced photocatalytic H2 evolution[J]. ChemCatChem, 2021, 13(5): 1355-1361. doi: 10.1002/cctc.202001847 [11] LUO M, LIU Y, HU J C, et al. One-pot synthesis of CdS and Ni-doped CdS hollow spheres with enhanced photocatalytic activity and durability[J]. ACS Applied Materials & Interfaces, 2012, 4(3): 1813-1821. [12] ZHU M S, OSAKADA Y, KIM S, et al. Black phosphorus: A promising two dimensional visible and near-infrared-activated photocatalyst for hydrogen evolution[J]. Applied Catalysis B: Environmental, 2017, 217: 285-292. doi: 10.1016/j.apcatb.2017.06.002 [13] HABIBI M H, RAHMATI M H. Fabrication and characterization of ZnO@CdS core-shell nanostructure using acetate precursors: XRD, FESEM, DRS, FTIR studies and effects of cadmium ion concentration on band gap[J]. Spectrochimica Acta. Part A, Molecular and Biomolecular Spectroscopy, 2014, 133: 13-18. doi: 10.1016/j.saa.2014.04.110 [14] 胡建民, 王蕊, 王春婷, 等. 晶体X射线衍射模型和布拉格方程的一般推导[J]. 大学物理, 2015, 34(3): 1-2. HU J M, WANG R, WANG C T, et al. X ray diffraction model of crystal and general derivation of Bragg equation[J]. College Physics, 2015, 34(3): 1-2 (in Chinese).
[15] HOTA G, IDAGE S, KHILAR K. Characterization of nano-sized CdS–Ag2S core-shell nanoparticles using XPS technique[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2007, 293: 5-12. [16] ABE T, KASHIWABA Y, BABA M, et al. XPS analysis of p-type Cu-doped CdS thin films[J]. Applied Surface Science, 2001, 175/176: 549-554. doi: 10.1016/S0169-4332(01)00147-7 [17] NING X F, LU G X. Photocorrosion inhibition of CdS-based catalysts for photocatalytic overall water splitting[J]. Nanoscale, 2020, 12(3): 1213-1223. doi: 10.1039/C9NR09183A [18] GUAN L, CHEN X B. Photoexcited charge transport and accumulation in anatase TiO2[J]. ACS Applied Energy Materials, 2018, 1(8): 4313-4320. doi: 10.1021/acsaem.8b00944 [19] XIA T, LI N, ZHANG Y L, et al. Directional heat dissipation across the interface in anatase-rutile nanocomposites[J]. ACS Applied Materials & Interfaces, 2013, 5(20): 9883-9890. [20] TAUC J, GRIGOROVICI R, VANCU A. Optical properties and electronic structure of amorphous germanium[J]. Physica Status Solidi (b), 1966, 15(2): 627-637. doi: 10.1002/pssb.19660150224 [21] MATSUMOTO Y. Energy positions of oxide semiconductors and photocatalysis with iron complex oxides[J]. Journal of Solid State Chemistry, 1996, 126(2): 227-234. doi: 10.1006/jssc.1996.0333 [22] TRASATTI S. The absolute electrode potential: An explanatory note (Recommendations 1986)[J]. Pure and Applied Chemistry, 1986, 58(7): 955-966. doi: 10.1351/pac198658070955 [23] ZHOU H L, QU Y Q, ZEID T, et al. Towards highly efficient photocatalysts using semiconductor nanoarchitectures[J]. Energy & Environmental Science, 2012, 5(5): 6732-6743. -
 点击查看大图
点击查看大图
计量
- 文章访问数: 452
- HTML全文浏览数: 452
- PDF下载数: 2
- 施引文献: 0
