

-
随着科学技术的迅速发展,化学品的数量急剧增多;2019年5月至2021年7月的两年间,美国化学文摘(Chemical Abstracts Service,简称CAS)登记的化学品数目就从1.5亿增长至2.5亿(http:/www.cas.org),两年注册量达1亿. 数量巨大、种类繁多的化学品进入环境中,会在不同的环境介质中产生相应的物理、化学和生物过程,部分化学品可能会引发巨大的生态风险与健康风险,而这些高风险污染物的环境行为已成为环境科学领域关注的重要问题[1–5];污染物与不同环境界面的相互作用机制更是其中的核心研究内容之一[6]. 污染物经各种暴露途径进入到环境后,会在水、大气、土壤等不同环境介质中和介质之间进行迁移转化. 事实上,无论是不同环境介质间还是在单一环境介质中,均存在不同类型的环境界面. 环境化学品在这些环境界面上能够发生吸附-解吸、氧化还原、催化降解乃至转化生成毒性更强的污染物等环境化学过程[7]. 因此,解析污染物的环境界面行为机制至关重要[8]. 遗憾的是,单纯依赖实验研究不仅难以满足大量化学品检测的需求,而且即便是借助同步辐射等先进大型科学装置,亦难以完全解析污染物在不同环境界面吸附、生成、转化等行为的分子机制[9-10]. 而采用环境计算化学手段,则有望通过相应的模拟分析揭示污染物界面行为的结构基础和化学机理,在污染物界面相关行为机制探索方面均展示出独特魅力[11].
污染物界面作用模拟中普遍使用的典型分子间作用方法主要有量子力学(quantum mechanics,简称QM)、分子动力学(molecular dynamics,简称MD)模拟及基于粗粒化模型(coarse-grained model,简称CG模型)的粗粒化分子动力学(coarse-grained molecular dynamics,简称CG-MD)模拟和耗散粒子动力学(dissipative particle dynamics,简称DPD)模拟等. 由于不同环境界面问题关注的尺度和化学性质的不同,其所适用的计算方法各不相同(图1). 如QM方法虽能够在原子水平上提供传统化学键生成和断裂的信息,对涉及分子结构变化的污染物界面生成和转化机制研究方面有很好的应用效果,但因其计算量庞大,故只适用于处理有限原子数目的体系[12]. 全原子MD模拟方法能够在微观水平上解析污染物在不同环境介质中的动态作用机制,相较于QM方法,其适用的时间尺度更长,空间尺度也更大[13]. 基于粗粒化模型的CG-MD和DPD方法能够在介观尺度上研究污染物的环境界面行为,适用于更加复杂而宏观的环境体系,但模拟的精度不如MD模拟[14-15].
针对不同的环境界面问题,需要选择不同的计算模拟方法去平衡计算精度与所需资源. 随着计算机运算能力的大幅提升,通过建立不同尺度的污染物-环境界面作用模型,使用分子间相互作用计算模拟方法解析污染物形成、吸附、扩散乃至转化的环境化学机制,同时实现不依赖实验数据的污染物环境界面行为的高通量解析成为可能. 近年来,计算模拟方法在污染物界面行为特征以及均相环境转化路径等研究方面均取得了诸多进展,在辅助揭示污染物环境行为化学机制的同时,为进一步评估污染物的环境毒性和生态风险提供了高效的评估工具[16-17]. 但是,污染物种类的多样性及环境体系的复杂性仍然对通过计算方法解析污染物环境界面行为的微观机制提出了更高要求. 对于复杂的环境系统而言,污染物在其中的环境界面行为涉及微观到介观多个尺度. 根据研究环境界面问题的特点和研究目标,选用多种计算方法联用,从不同的视角提供多样化的机制信息正在成为必然[18-19]. 本文对环境化学领域应用较多的3类计算模拟方法及它们在研究污染物环境界面行为方面的应用进行了回顾与总结,评述了不同计算模拟方法在处理不同尺度环境问题时的适用性,并对未来的发展作了展望.
-
量子化学计算是目前解析化学物质各种物理化学性质的最精确的方法. 早在1927年,科学家首次利用量子力学阐述了最简单的同核双原子分子氢气中两个氢原子间化学键的形成过程,标志着量子化学的诞生. 量化计算的核心涉及多体薛定谔方程,而目前仍无法精确求解薛定谔方程,需要运用一定的近似方法进行求解. 因此,多种旨在兼顾精确性与计算量的近似方法不断涌现,而其中组态相互作用、多体微扰理论、密度泛函理论(density functional theory,简称DFT)及各种各样的半经验方法均很好地实现了化学结构的定量描述[20]. 随着方法学的不断进步,量子化学计算已经被广泛引入环境、医药、生物、材料等领域用于解决相关科学问题[21–24]. 其中,DFT是一个通过电子密度分布研究多电子体系波函数的方法,在兼顾精度的同时极大地节约了计算量,成为当前量子化学的主流算法. DFT方法根植于Hohenberg-Kohn定理,基于定理一可将薛定谔方程中的基态能量表示为基态电子密度的泛函. 通过这一理解可将一个N电子体系薛定谔方程中的3N个自由度简化为3个,即动能项、外势能项和相互作用项,极大地降低了运算复杂度. 根据Hohenberg-Kohn第二定理,能量最低的电子密度分布方式就是薛定谔方程的基态解[25]. 进一步,DFT对总能量进行了近似和简化,并将其误差项作为交换-关联能. 因此,体系总能量即为动能、外势能、相互作用能三项与交换-关联能之和. 对交换-关联能的计算目前主要包括局域密度近似、广义梯度近似、含动能密度的广义梯度近似、杂化泛函等方法[26]. 此外,第一性原理计算也不时用于环境领域问题的计算模拟. 严格意义上的第一性原理计算是从求解最基本的QM公式薛定谔方程出发,不引入或尽量少地引入经验参数的基于非经验参数的理论方法,因此又被称为从头算方法[27]. 目前,广义的第一性原理计算更多是泛指通过QM手段研究周期性体系的量化计算方法. 常见的量子化学程序除了Gaussian、VASP、Materials Studio中的DMol3和CASTEP模块、Turbomole等付费工具以外,对学术用户免费的ORCA、xtb、CP2K等日益受到学者的关注.
-
随着环境问题所需计算尺度的增加,若继续采用量子化学计算方法,则需要占用海量的计算资源. 考虑到量子效应随着问题尺度增大而减弱乃至消失,其运动趋向于遵循宏观动力学规律与统计学分布. 当所研究的问题不涉及传统化学键生成和断裂的污染物界面行为时,MD模拟将是更合适的计算方法. MD模拟通过求解经典牛顿力学方程,得到体系内各原子在不同时刻的位置和速度,得到分子的运动轨迹,以此来描述体系的动态变化过程. 分子在系统中的总势能是体系内分子中各原子位置的函数U(r),因此质量为mi的某原子i所受力Fi为:
根据牛顿力学定律,若vi代表该原子的速度矢量,ri代表其位置矢量,在该位置处的原子i此刻的加速度为:
$ \mathrm{\Delta }t $ 时间间隔后,该原子的速度和位置分别为:其中,
${\boldsymbol{v}}_{\boldsymbol{i}}\left(t\right)$ 、${\boldsymbol{v}}_{\boldsymbol{i}}(t+\mathrm{\Delta }t)$ 分别为t时刻和$ t+\mathrm{\Delta }t $ 时刻原子的速度,${\boldsymbol{r}}_{\boldsymbol{i}}\left(t\right)$ 和${\boldsymbol{r}}_{\boldsymbol{i}}(t+\mathrm{\Delta }t)$ 分别为t时刻和$ t+\mathrm{\Delta }t $ 时刻原子位置,${\boldsymbol{F}}_{\boldsymbol{i}}\left(\boldsymbol{t}\right)$ 为原子t时刻所受的力[28].相对于量子力学对于分子结构和原子间相互作用的描述,在遵循分子力学方法的MD中,分子力场的合理选择尤其重要. 由原子类型、势函数与力常数构成基本要素的分子力场将原子间作用看成类似弹簧的弹性力,用简单的数学函数描述原子间作用,其力场参数主要来源于实验值及高精度量子化学计算. 针对不同应用体系,常需要选择不同的力场进行MD模拟(表1). 譬如,如针对材料体系的COMPASS力场,针对蛋白质体系的ff19SB力场,针对磷脂体系的lipid21力场,针对核酸体系的OL21力场,针对有机分子的GAFF力场等[29]. 最常用的MD模拟程序有GROMACS、Lammps、AMBER、Materials Studio中Forcite模块、NAMD等.
-
随着环境问题尺度进一步增大到介观范围,体系的原子数将急剧增加,常规MD模拟受到计算资源限制,很难适用于具备如此多自由度的介观尺度体系. 为了应对这种情况,可以把研究体系中分子或分子的某一部分近似看成一个整体,从而忽略其内部的键长、键角、二面角等信息. 即该部分原子簇被处理为没有内部结构的珠子,在计算中只考虑珠子之间的作用以保留体系的基本化学性质. 这样,在牺牲一定精确度的情况下极大地减轻了计算量,从而能扩大模拟时空尺度到至微秒和微米级别. 这种突破全原子建模尺寸的模型称为CG模型[30]. 与全原子MD相似,可以对CG模型进行MD模拟,由于CG体系具有更少的自由度和较软的相互作用势,CG-MD模拟能够获取污染物在复杂环境界面中更长时间的动态变化信息[31]. 除了基于MARTINI力场等具有多尺度穿越性的CG-MD方法[32],最早由Hoogerbrugge和Kopelman于1992年提出并应用于微观水动力现象研究中的DPD是一种很适合于污染物复杂界面行为研究的基于CG模型的典型计算模拟方法[33]. 1994年,Kong等将“珠子-弹簧模型”(Bead-Spring Model)引入DPD方法中,进一步拓展了这种方法的使用范围[34]. 1995年,Español通过将“涨落-耗散定律”与DPD方法结合,重新推导了新算法,并将保守力引入体系中[35]. 1997年Groot和Warren揭示了DPD方法中的保守力参数与Flory-Huggins参数之间的关系,促使DPD模型更贴近模拟体系真实情况,为其多领域应用提供了可能[36].
在实际问题的DPD应用研究中,根据需要将分子中的特定原子簇视为一个珠子,各珠子间所受的力、位置、速度与时间的关系依然遵循牛顿力学定律. 对于一DPD定义的珠子i而言,有:
其中,该珠子所受的力Fi来自其与其他粒子j之间的保守力
${\boldsymbol{F}}_{ij}^{\mathrm{C}}$ (conservative force)、耗散力${\boldsymbol{F}}_{ij}^{\mathrm{D}}$ (dissipative force)和随机力${\boldsymbol{F}}_{ij}^{\mathrm{R}}$ (random force)三大相互作用(图2):这3种珠子间相互作用力可分别表示为:
其中,
$ {a}_{ij} $ 为保守力参数,用以衡量珠子间相互作用强度.$ \zeta $ 为耗散系数,$ \sigma $ 为随机力强度,$ {\xi }_{ij} $ 为符合高斯分布的随机函数.${\omega }^{\mathrm{C}}\left({r}_{ij}\right)\mathrm{、}{\omega }^{\mathrm{D}}\left({r}_{ij}\right)\mathrm{、}{\omega }^{\mathrm{R}}\left({r}_{ij}\right)$ 均为权重因子,是珠子i和j间距离$ {r}_{ij} $ 的函数,而$ {r}_{C} $ 为系统的截断半径[36]:粗粒化模型及相应的计算模拟方法在高分子、生物大分子等具有软物质特征的体系及石油化工等工程领域亦有广泛的应用[15,31-32]. 能够用于CG-MD和DPD计算模拟的程序有Lammps和Materials Studio中的Mesocite模块等.
-
QM方法不仅能够描述污染物与环境介质乃至界面分子间的相互作用,评估污染物在不同环境界面的吸附自由能等热力学信息,还能描述界面反应过程化学键的生成和断裂,获取关键界面化学反应的活化能、焓变等热力学和动力学数据,解析污染物在环境界面生成、转化过程中的中间体和过渡态,为揭示污染物的环境赋存状态和界面生成转化过程微观机制提供方法支撑.
基于量化计算,可以准确获取污染物在不同环境表面吸附过程的分子机制及热力学信息. 天然矿物和有机质是沉积物和土壤的重要固相组成成分,它们与污染物间的相互作用影响着污染物进入土壤后的归趋和转化行为. 甲基汞的生物有效性和环境移动性深受具有四边形结构的层状硫化铁矿物mackinawite(FeS)的表面反应控制,Guo等[37]基于色散校正的密度泛函理论对甲基汞在黑烟硫铁矿(001)、(011)和(111)等不同晶面上的吸附位点及吸附强度开展了系统的第一性原理计算,为从分子水平上理解甲基汞在其表面吸附的位面选择和键合机制提供理论依据. 分析结果表明,甲基汞优先与FeS表面的Fe位点结合而非S位点结合. 也正因为这一差异,甲基汞在FeS(001)晶面上的吸附强度弱于FeS(011)和FeS(111)两个晶面,FeS(111)面上的吸附作用最强. 前者界面最稳定构型是单齿S-Hg配合物,而甲基汞在FeS(011)和(111)面最稳定吸附构型分别是单齿Fe-Hg配合物和二齿Fe-Hg-Fe配合物. 基于量化计算解析的黑烟硫铁矿不同晶面对甲基汞的差异化吸附机制将有助环境学家进一步优化黑烟硫铁矿清除地下水和土壤系统中甲基汞的治理方案. Ahmed等[38]通过QM计算与实验相结合的研究策略得到了典型的抗生素类污染物磺胺与土壤有机质(soil organic matter,简称SOM)间的相互作用机理,结果表明二者的作用强度主要依赖于SOM的化学组成而非其含量. 虽然磺胺可以分别通过自身的极性基团和芳环与SOM中对应的极性和疏水位点主要通过氢键和π-π相互作用进行结合,但是亲水的磺胺仍更倾向于吸附在SOM极性位点上. 正因如此,干燥土壤中SOM组分对磺胺的亲和力呈阳离子>阴离子>肽>碳水化合物>酚和木质素单体>木质素二聚体>含氮杂环化合物>脂肪酸>甾醇>烷基化芳香族化合物>脂质、烷烃和烯烃的递降趋势,而土壤含水率的升高会使得可与磺胺结合的位点减少,而大大弱化SOM对磺胺的固定作用;进一步的定量构效模型证实SOM组分的偶极矩和原子电荷是定量评估其磺胺吸附性能的重要结构描述符. 此外,量化计算同样广泛应用于污染物与环境纳米材料的作用机制研究中. Li等[39]采用石墨烯作为磁性还原氧化石墨烯的简化模型,基于色散校正的DFT计算和实验测定推测了4-庚基苯酚、4-叔辛基苯酚和4-壬基酚等3种典型烷基酚在材料表面气相吸附的构型和可能机制. 作者尝试了垂直和平行吸附两种构型,发现三种烷基酚平行构型的吸附作用均强于其相应垂直构型,且4-壬基酚的材料亲和力高于其他两种烷基酚,这与实验测定相符. 但实验显示水中的4-叔辛基苯酚在相同条件下比4-庚基苯酚更难以被磁性还原氧化石墨烯吸附去除,而吸附能计算值的结论恰好相反. 鉴于4-庚基苯酚气相中伸展的烷基链在水溶液中会发生卷曲以减少其与极性水分子接触的面积,而4-叔辛基苯酚可预见的液相和气相构型差异远低于4-庚基苯酚,这一简化计算和实验不相符合之处应部分源于未将溶剂化效应等考虑在内有关. 此外,计算显示典型烷基酚与环境材料间非均相多层物理吸附的驱动力是界面π-π、氢键和疏水作用. Zou等[40]使用M05-2X泛函计算了环己烷、苯衍生物和多环芳烃在(8, 0)锯齿型单壁碳纳米管上的吸附能,发现水相中的吸附能低于气相吸附能,说明在热力学上水相中材料对污染物的吸附比气相有利,且DFT计算得到的吸附能与实验值具有很好的一致性. 此外,计算研究还量化了π-π相互作用和硝基对这一作用的增强效应在污染物单壁碳纳米管表面吸附中的贡献. 通过比对环己烷及与其具有相同疏水性的假设芳烃的吸附能,可知污染物分子中的苯环和单壁碳纳米管苯环间的π-π作用对吸附能贡献了24%;而芳环上的硝基取代则由于其强吸电子效应进一步增强了污染物与单壁碳纳米管分子间π-π相互作用.
污染物在不同环境界面的生成、转化和降解的微观机制解析受到实验手段方法学的限制,QM方法为解决这一问题提供了有效手段. 在大气、土壤乃至水环境中存在各种不同性质和反应活性的气-固、液-固等复杂界面,而污染物在这些界面处发生的非均相生成转化是其至关重要的环境化学行为之一. QM方法在污染物环境非均相生成转化机制解析研究中发挥了重要作用. 譬如二噁英类物质和环境持久性自由基均为典型的环境有机污染物,了解其在大气中的非均相生成机制对于评估其环境风险极其重要. Mosallanejad等[41]首先基于量化计算分析了2-氯酚分子在纯净和羟基化二氧化硅表面进行化学吸附形成吸附态2-氯苯氧基的分子历程,结果显示相比羟基化表面需脱除水分子形成2-氯苯氧基自由基的141.8 kJ·mol−1高能垒,2-氯酚与二氧化硅形成的范德华络合物中酚羟基的H迁移到洁净表面O缺陷位点上只需31.8 kJ·mol−1的适度反应势垒. 随后采用Eley-Rideal机制对2-氯酚与吸附态2-氯苯氧基自由基的反应途径进行模拟,证实了二氧化硅对这一过程的催化作用,且反应分别经HCl或Cl2消除可生成氯代二苯并-对-二恶英或氯代二苯并呋喃中间体. 这一计算所得反应历程与高温实验结果相符,说明即使没有过渡金属参与,飞灰颗粒也会催化氯酚类物质生成二噁英. Pan等[42]揭示了大气中五氯酚在Fe(Ⅲ)-蒙脱石表面通过铁原子的3d空轨道和酚氧基团氧原子2p轨道相互作用形成表面负载的酚氧型自由基的转化机制,并指出Ca(Ⅱ)-蒙脱石和Na(Ⅰ)-蒙脱石表面的五氯酚因不存在类似的电荷转移而相对稳定. Assaf等[43]基于第一性原理计算模拟了燃烧冷却阶段苯酚在氧化铝表面生成酚氧型自由基的转化历程,计算得到苯酚分子在非羟基化的α-Al2O3(0001)面转化生成酚氧型自由基的活化能为48 kJ·mol−1. 量化计算除了在大气CuO[44]、α-Fe2O3[45]、Cu/Fe及其部分氧化构型[46]等颗粒表面的环境持久性自由基生成机制研究中有所应用以外,还在其与环境大气共存物质的反应机制解析中发挥重要作用. Pan等[47]通过DFT计算研究了大气中的O2、•OH和NO3自由基对Cu(Ⅱ)O表面负载的酚氧型自由基的作用机制,结果表明大气中的化学活性物质•OH和NO3会与表面负载酚氧型自由基发生反应,而大量存在的O2基态下则不会导致Cu(Ⅱ)O表面负载酚氧型自由基化学形态的转变. 此外,QM方法亦是解析污水处理等环境治理技术中污染物去除机理的有效手段. Liu等[48]使用实验和量化计算相结合的方法对刻面定制TiO2阳极材料废水电化学处理机制开展了研究,指出相比分散在水溶液中的游离态•OH,氧化对位取代酚类污染物的主要是表面负载的•OH和TiO2/Ti电极上直接的电子转移. 而对阳极酚氧化过程中锐钛矿TiO2高能(001)晶面和低能(101)晶面水吸附和解离的量化计算进一步表明,表面结合•OH介导的对位取代酚类氧化归因于形状定制的TiO2和水分子以及活性•OH之间的表面键合作用;高能(001)极性面之所以在酚类氧化上优于低能(101)面,源于其具有大量悬空键的低配位表面原子的高密度原子台阶、边缘和扭结. 这些微观机制的解析为电化学水处理环境技术研发高效TiO2基阳极材料提供了宝贵的理论经验. 类似地,Huang等[49]结合实验和DFT理论计算成功解析了基于过渡金属氧化物CuO的过硫酸盐活化过程机理,指出在CuO表面原位形成的表面负载苯氧基自由基有效触发过硫酸盐活化以降解污染物苯酚. 具体地说,这一表面负载苯氧基自由基的形成是涉及有机底物苯酚化学吸附和随后苯酚向CuO自发电子转移的一个内球过程,最终苯酚分别被过硫酸盐分子和表面结合的
${\rm{SO}}_4^{\cdot -}$ 通过非自由基和自由基途径氧化. QM方法在选择性催化还原法去除烟气中NOx的机制解析方面亦取得成功. 2021年Gao等[50]结合实验和DFT计算明确提出在α-Fe2O3(012)表面NH3选择性催化还原NO的反应路径不同于已知的NH3活化机理. 在这一催化体系中,NO优先以亚硝酰形态吸附在催化剂表面的三配位氧O3c位点,NH3则优先吸附在Fe5c位点. 一般认为催化反应是基于NH3活化机理进行,即吸附于Fe5c位点NH3先活化为-NH2物质,再与从O3c位点解吸的NO反应生成中间体亚硝酰胺NH2NO. Guo等发现热力学上更有利的反应历程应是以亚硝酰形态吸附在催化剂表面三配位氧O3c位点的NO迁移至五配位不饱和的铁阳离子Fe5c位点,与优先吸附在该位点的NH3相互作用生成过渡态NO···NH3,而后其中的NH3将一个H原子转移到相邻O3c位点形成中间体NH2NO和一个势垒为1.22 eV的O3cH基团. NH2NOH最终转化为N2和H2O则需要O3c位点辅助的一系列H转移和异构化反应;催化剂则通过与O2反应脱除表面O3cH完成再生. 其中,NO的活化及中间产物NH2NO的生成是整个催化循环的决速步骤. 这一NO活化机理的提出为开发高效的选择性催化还原催化剂提供了新的视角. -
当研究的界面体系更复杂,涉及的分子更多、体系更大时,选用QM方法需要耗费大量的时间和计算资源. 若研究的环境界面问题不涉及化学反应过程,则计算量更小且计算时间更短的MD模拟更加合适. MD模拟能够在原子水平下解析污染物与不同环境界面的动态作用机制,对评价、认知污染物的环境界面动态行为特征有重要意义. 目前,MD模拟已广泛用于研究污染物在含无机矿物、天然有机质、各类环境功能材料等多种环境界面体系行为的分子机制.
土壤矿物及其孔洞的存在对于多环芳烃等环境污染物的土壤滞留具有重要作用. 2016年Sui等[51]采用全原子MD及机器学习相结合的方法对16种多环芳烃混合物的SiO2纳米孔吸附作用进行了研究. 模拟结果提示污染物与SiO2纳米孔内表面的相互作用主要是吸附能贡献超70%的范德华力,且由于缺乏可用表面和自由体积,因此疏水多环芳烃混合物在SiO2纳米孔中的吸附主要受其分子扩散影响而非竞争吸附机制控制. 改变混合物中单个多环芳烃的浓度比和混合物中多环芳烃的性质,可改变竞争吸附和分子扩散在其中的比重. 矿物内表面对污染物的滞留作用亦不容给忽视,早在2010年Aristilde等[52]就通过MD模拟了氧四环素与Na(Ⅰ)-蒙脱石内部微界面的相互作用,结果表明Na(Ⅰ)-蒙脱石的层状结构间距和结构电荷定位均会影响嵌入其硅氧四面体和铝氧八面体片层间氧四环素的结合构象. 矿物表面的吸附是控制铀(Ⅵ)环境迁移的关键因素. Kerisit等[53]2014年选用正长石(001)面和高岭石(001)氧八面体片,分别模拟了水环境下铀(Ⅵ)的两类中性铝硅酸盐界面吸附行为. MD模拟结果显示
$\rm UO_2^{2+} $ 在所研究的两种表面都以双齿内球表面络合物的形式吸附,但由于两个界面官能团的差异和正长石在$\rm UO_2^{2+} $ 吸附时释放表面钾离子的能力,使得$\rm UO_2^{2+} $ 在正长石的界面吸附更稳定. 考虑到大量存在的碳酸盐,该研究还进一步模拟了$\rm UO_2^{2+} $ 与最多3个碳酸根配体形成的配合物中性铝硅酸盐表面的吸附,计算结果提示铀酰配合物UO2CO3和$\rm UO_2(CO_3)_2^{2-}$ 在正长石表面上是稳定的,但是高岭石则不然. Willemsen等[54]2019年采用元动力学这一增强采样的方法,基于MD模拟分析了水环境中6种邻苯二甲酸酯与Ca(Ⅱ)-蒙脱石的相互作用,证实这类具有内分泌干扰活性的化学增塑剂不仅能在Ca(Ⅱ)-蒙脱石外表面吸附,还可存在于层间纳米孔中;模拟估算的分配系数与实验值吻合. 计算显示邻苯二甲酸酯的表面吸附主要占据强疏水的不带电区域,以平行吸附的构型为主,且其吸附程度和黏土表面电荷密度间存在很强的反比关系,而亲和力是由小的有利范德华贡献和大的有利熵贡献引起.环境土壤有机质组成多样、结构复杂,在微生物活动、土壤颗粒聚集、植物生长和碳储存等多个环境生物地球化学过程中发挥着重要作用;其对污染物吸附作用显著影响着污染物进入环境后的迁移行为乃至归宿. 为更好地评价溢油的整体环境风险,2015年Zhu等[55]用甲苯来模拟溢油,研究了水和沉积物中的腐殖酸在溢油发生时对重油沥青聚集的影响,通过全原子MD分别分析了甲苯/水和纯水环境中沥青与风化褐煤腐殖酸间的共聚集作用,以期解析沥青从油到水的传输行为. 结果显示在甲苯/水体系,腐殖酸会在甲苯-水界面处聚集形成腐殖酸层并在邻近水相一侧与水形成氢键,沥青则是在这一腐殖酸层邻近甲苯这一侧以平行或T形堆积方式聚集. 在纯水环境中,平行堆积的沥青质经过表面吸附和结构变形而隔离在腐殖酸分子聚集体形成的疏水内腔中. 比较甲苯-水和纯水环境中腐殖酸对沥青聚集的影响,可知腐殖酸对于后者的影响更为显著. 2020年Petrov等[56]采用全原子MD与微扰自由能方法相结合的技术对不同类型有机分子在水活度接近0和1两种不同水合水平标准风化褐煤腐殖酸体系的界面行为进行了模拟分析,计算得到的有机物的水合自由能及其在典型土壤有机质表面的吸附自由能与实验证据具相关性,且能在原子水平上揭示有机分子与SOM分子及水分子间的相互作用. Vialykh等[57]通过MD模拟分析了氢键、亲水/疏水表面积和π-π堆积相互作用在腐殖质组成分子聚集过程以及环境污染物的吸附中所起的作用. 研究显示在具有小分子的高度亲水腐殖质模型和具有较大分子片段的高芳香性和疏水性腐殖质模型中,驱动组成分子聚集的主导非共价作用是不同的,前者为氢键,后者源于疏水和π-π堆积. 同样地,不同性质的污染物与腐殖质的相互作用也呈差异化的分子机制. 极性污染物苯酚通过氢键与腐殖酸相互作用,而非极性污染物苯则是通过疏水和π-π堆积与腐殖酸作用. 此外,非极性污染物还会引起腐殖酸组分聚集体内部分子的结构重排形成内部疏水空间,而这一疏水空间可捕获非极性污染物并形成在热力学上更加稳定的污染物-腐殖酸复合物结构,增强其在腐殖酸中的滞留时间. Tunega等[58]则以羧基和脂肪链分别构建腐殖酸纳米孔的极性和非极性结构,模拟了极性污染物2-甲基-4-氯苯氧乙酸和非极性污染物萘在腐殖酸纳米孔结构中的吸附过程. 结果显示不同极性的污染物吸附强度相似,但吸附机制不同. 极性羧酸倾向于在亲水的水-腐殖酸界面处聚集稳定,而非极性萘则被吸附至纳米孔结构的疏水内部. 2020年Zhao等[59]通过全原子MD和静电势表面模拟研究了石英表面、标准风化褐煤腐殖酸和水对多环芳烃吸附的贡献. 模拟结果表明,多环芳烃的腐殖酸吸附性能远高于其在石英表面的吸附作用,且多环芳烃与风化褐煤腐殖酸间的主要作用力不是π-π堆积,而是污染物与腐殖酸中羧基和羟基的相互吸引. 水分子的存在干扰了疏水污染物的表面吸附,且其在石英表面的迁移率远高于腐殖酸.
污染物在纳米尺度环境功能材料表面的吸附能够影响其在环境中的迁移转化及毒性. 譬如典型环境纳米材料石墨烯及其改性产物因具有较高的比表面积和物化性质,已被用于污染治理技术研发中. 林等[60]研究了双酚A在含氧化石墨烯的水溶液中的吸附过程,结果表明双酚A与氧化石墨烯间的π-π相互作用是吸附主导作用,而静电和氢键作用可增强材料对污染物的吸附能力. Wang等[61]通过50 ns的MD模拟,计算得到了43种中性芳香类污染物与石墨烯和具有羟基、环氧和羰基等不同官能团修饰的氧化石墨烯相互作用的模式. 发现石墨烯表面对这些中性芳香族化合物的亲和力比氧化石墨烯强,表面具有可解离羟基和羰基的氧化石墨烯与污染物间可形成氢键,而无法解离的环氧基修饰的表面则不可以. 研究构建的吸附平衡系数K理论线性溶剂化能相关预测模型进一步证实氢键给体能力指标εa与色散/疏水作用指标V对石墨烯的中性芳香族化合物吸附能力lgK影响显著,而氧化石墨烯则主要受εa控制,与MD所获得界面作用分析机制相符合. Tang等[62-64]基于GROMACS实现全原子OPLS力场来成功模拟了可离子化芳香族有机物等多种污染物在氧化石墨烯表面的吸附过程, 并得到了与实验观测相吻合的结果. 赵超锋等[65]同样使用Jorgensen等开发的全原子OPLS力场构建了氧化石墨烯和苯酚、α-萘酚和4-辛基酚的结构并用于MD模拟,通过伞形采样计算了污染物的吸附能. 该研究发现无论是直接吸附还是通过形成分子团簇的间接性吸附,疏水作用都是3种酚类有机污染物在氧化石墨烯表面吸附的主导作用力,而三者共存时的竞争吸附中静电相互作用的影响相对增强. MD模拟在新污染物的环境界面行为评估方面亦有所应用. 以在海洋、淡水、土壤等环境广泛检出的微纳塑料(micro- and nanoplastics,简称MNPs)为例,具有大比表面积的MNPs不仅能本身自发聚集形成具有一定排列方式的纳米团簇,还可作为新的环境微界面影响其它污染物环境赋存形态、水平乃至归趋[66-67]. 考虑到MNPs吸附污染物的能力与其环境赋存粒径等有关. Ramalho等[68]通过分子动力学模拟分析了聚乙烯、聚对苯二甲酸乙二醇酯和聚酰胺尼龙6等3种常见材质塑料纳米粒子的水环境聚集行为,发现虽然三者均会快速自发地聚集成纳米团簇,但其自组装和聚集模式与它们各自分子结构特征密切相关. 如聚乙烯纳米团簇更加紧凑有序,而聚对苯二甲酸乙二醇酯弯曲的链形成纠缠堆积结构,其中芳环优先以平行有序的方式取向. 2020年Guo等[69]通过MD模拟揭示了聚酰胺、聚苯乙烯和聚丙烯对Sr2+的吸附以静电相互作用为主导;2021年Wang课题组[70-71]对运用MD模拟研究了水环境中典型农药和抗生素在聚乙烯MNPs上的吸附作用,证实微塑料是农药和抗生素的重要环境载体. 他们采用巨正则蒙特卡洛模拟和MD计算分析了吡虫啉、苯醚甲环唑和噻嗪酮等3种农药的聚乙烯MNPs界面行为,发现农药分子与微塑料C、H原子之间存在复杂的相互作用;而MD模拟显示范德华力等非键相互作用是水溶液中聚乙烯MNPs表面吸附盐酸四环素、盐酸金霉素和盐酸土霉素的主要相互作用,且聚乙烯MNPs对盐酸金霉素和盐酸土霉素的亲和力最高,盐酸四环素次之.
-
CG-MD和DPD方法是计算微观和介观尺度体系行为的重要方法,能够半定量地给出在介观尺度上污染物在复杂环境界面存在下的存在状态和扩散行为等信息,为评估污染物在环境中的迁移转化提供理论支撑. 以在医药、化妆品、催化等领域广泛应用的人工纳米颗粒(Nanoparticiles,简称NPs)为例,其进入环境后的存在形态、分布乃至生态效应已受到广泛关注[72]. 早在2015年Lado Touriño等[73]就基于MARTINI力场建立了水和碳纳米管CG模型,并通过CG-MD计算模拟说明了水环境中碳纳米管对水分子扩散行为的影响,结果表明不同长度和浓度的碳纳米管对水扩散系数的降低程度不同,并证明了水分子在碳纳米管内部的扩散具有各向异性. 但是,不同于全原子模型,将几个水分子当作一个基本粒子的CG模型难以支持外部环境水分子各向异性扩散的模拟. 2018年Wang等[74]对包括2种金属、7种金属氧化物和3种碳基NPs在内的12种NPs分别在纯水(pH 3.0)和藻类介质(pH 8.0)且有腐殖质类似物存在下的聚集和分散行为进行了比较系统的DPD模拟,结果表明腐殖质对特定NPs稳定性的影响均存在一个临界浓度. 在环境腐殖质浓度低于临界浓度时,腐殖质浓度增加,NPs的稳定性和分散性随之增加. 但是在临界浓度以上时,水中腐殖质含量的增加反而使得NPs不稳定且更趋于聚集. 从相互作用性质而言,在腐殖质存在的情况下,疏水和氢键相互作用主要控制NPs的聚集,而静电相互作用和空间排斥促使纳米粒子在水相分散. 2021年Dettmann等[75]以内表面修饰不同疏水和亲水官能团的碳纳米管模拟土壤环境疏水性构件土壤有机质的表面和限制效应,用聚乙烯、聚丙烯、聚苯乙烯和聚环氧乙烷等材质聚合物模拟MNPs,采用CG-MD模拟了水中典型MNPs与土壤有机质表面及功能化空腔内部的相互作用. 结果表明MNPs可以吸附在模拟的土壤有机质颗粒表面并被捕获在其空腔内部,且MNPs和土壤有机质结构的疏水性以及土壤有机质空腔/空隙的化学性质是控制土壤MNPs积累的重要参数. 鉴于MARTINI力场适用的粒子类型的限制以及土壤有机质构成的复杂性,2018年Feng等[76]基于Vienna Soil-Organic-Matter Modeler生成土壤有机质凝聚相模型,并开展一系列DPD模拟成功评估了全氟烷基磺酸和六氯丁二烯在水-土壤有机质体系的扩散系数. 结果表明全氟烷基磺酸的扩散系数随其碳链长度的增加而降低,且采用含水土壤有机质体系更接近于环境真实. DPD方法在沥青的油/水界面行为研究中亦有所应用. Ruiz-Morales等[77]构建了沥青的CG模型,其核心是多环芳烃,而周围是烷基. 采用甲苯作为油的模型分子,对沥青质在油/水界面的分子取向和赋存形态开展了DPD模拟. 结果表明,无论沥青分子在体系中的初始构型是平行于油/水界面、垂直于油/水界面或与油/水界面成45°倾斜角,最终均倾向于以其芳环平躺于油/水界面,而脂肪链深入油相且垂直于油/水界面的分子取向稳定. 提高沥青分子在油/水界面的表面覆盖率亦不会改变这一分子取向,但由于空间位阻的存在使得部分沥青整体迁移到体相,只余部分在界面处;整体进入到油相的沥青会通过其芳环发生π-π堆叠作用而聚集. 此外,烷基末端有无含氧官能团对沥青的界面行为影响显著. 末端含氧的烷基会指向油/水界面,含氧基团分布在油/水界面近水相处,而沥青的芳环反而进入油相中.
-
Martin Karplus、Michael Levitt和Arieh Warshel三位科学家凭借为复杂化学系统创立了多尺度模型这一贡献获得2013年诺贝尔化学奖. 对于复杂的环境系统而言,污染物在其中的环境界面行为涉及微观到介观多个尺度. 而建立典型污染物参与的界面行为多尺度环境化学模型,逐级揭示污染物环境界面行为的化学机制亦不再遥远. 在实际环境界面问题的研究中,根据研究问题的性质和目标,采用不同的计算模拟方法,才有可能深入认识污染物环境界面行为的微观机制. 如2014年Ding等[78]结合DFT和MD模拟解析了二苯醚和9种不同溴化程度的多溴二苯醚在石墨烯表面的吸附行为. MD分析发现这类污染物在石墨烯表面吸附是一个主要由疏水作用驱动的快速物理吸附过程,而QM计算获得的电子态密度、电荷转移和热力学等信息表明这一界面作用强度随着溴化程度的增加而增加,且苯环上醚键邻位的溴取代能够通过空间位阻效应进而改变两个苯环的相对位置,并进一步对多溴二苯醚在石墨烯表面的吸附构型及稳定性产生显著影响. 2016年Cortés-Arriagada和Toro-Labbé[79]采用QM和MD相结合的方法研究了硅掺杂石墨烯对甲基砷的吸附去除机制. QM计算表明三价和五价甲基砷都是通过Si—O结合而与硅掺杂石墨烯表面形成稳定的化学吸附作用,随后MD分析显示在环境温度(300 K)下,中性pH水环境中的三价和五价甲基砷将分别主要以其中性和阴离子形式被硅掺杂石墨烯吸附去除. 2018年Tang等[64]结合实验、QM和MD揭示了芳香族化合物在氧化石墨烯的优先吸附位点. 结果显示芳香族化合物倾向于在氧化石墨烯褶皱和边缘处吸附,前者源于较高的原子和电子密度以及贡献了污染物迁移空间位阻的粗糙度,后者则主要受边缘附近较强的π-π相互作用和氧化石墨烯表面羧基与芳香族化合物间较强的氢键所驱动. 此外,水介导的空间位阻和氧化石墨烯表面羧基的柔韧性也有助于芳香族化合物的边缘吸附. 2021年Jiang等[19]结合DFT计算和MD模拟解析了纳米气泡对3种全氟烷基磺酸在石墨烯以及氨基和羟基改性石墨烯表面吸附的影响机理. 研究首先通过DFT计算得到了真空条件下全氟烷基磺酸在石墨烯表面的吸附能,结果表明磺酸基团与氨基间的强静电相互作用使得全氟烷基磺酸在氨基改性石墨烯表面吸附能最高,磺酸基团与羟基间的氢键作用则是污染物在羟基化石墨烯表面吸附的主要驱动力之一,而表面静电势趋于0的未改性石墨烯对全氟烷基磺酸的吸附能力最差. 有趣的是,MD模拟显示水分子的存在虽不会显著影响污染物与氨基化石墨烯的界面作用,但会与磺酸基团竞争表面羟基形成氢键作用,进而导致全氟烷基磺酸无法稳定吸附在羟基化石墨烯表面;C—F链的疏油疏水特性亦使得污染物难以在未修饰石墨烯表面稳定吸附. 纳米气泡的加入可以使得水溶液中的全氟污染物通过空气-水界面富集,碳氟链完全延伸到空气相. 但是,MD模拟显示纳米气泡的存在无法改善羟基化石墨烯对污染物的去除能力,只能分别通过附着在材料表面的纳米气泡和分散在溶液中的纳米气泡促进未修饰和表面氨基修饰石墨烯对全氟烷基磺酸的去除性能. 这一研究充分利用了QM和MD方法不同的特点,类似的计算方法联用应用于Feng等[18]对水溶液中全氟烷基磺酸在α-Fe2O3表面的吸附机制探索. 研究首先通过MD模拟发现水溶液中这些典型全氟烷基磺酸污染物在α-Fe2O3表面的吸附并非是传统的垂直构型,而是以碳链平行于矿物表面的方式进行吸附,且这一界面构型很可能是通过污染物长链中C—F键的F原子与羟基化α-Fe2O3表面H原子间形成的氢键所稳定的. 进一步在真空条件下开展QM计算,发现吸附源于F原子2p轨道和H原子1s轨道相互重叠所形成的分子间弱相互作用和磺酸基团与表面的静电相互作用. 不同方法在解决实际环境问题中的相互配合不仅表现在方法联用上,还表现在方法发展中,如第一性原理分子动力学(first principle molecular dynamics,简称FPMD)模拟方法就将电子结构计算和分子动力学分析相结合,既能提供共存环境组分的可能界面吸附情况,又可以在高计算精度水平模拟界面作用性质等,在一定程度上耦合了密度泛函理论计算及MD模拟的两者优势. 重金属层状硅酸盐的成核和沉淀是重金属污染的重要稳定化机制,而这一环节可能发生在其吸附到黏土边缘的过程中. 2019年Zhang等[80]以Ni2+为模型重金属阳离子,采用FPMD模拟了黏土边缘重金属层状硅酸盐的非均相成核过程. 计算分析显示在黏土边缘直接形成Ni层状硅酸盐的同步成核路径在热力学上比先生成Ni(OH)2再通过硅化作用转化为Ni层状硅酸盐的逐步成核路径更有利.
-
环境界面无处不在,污染物环境界面化学机制一直是环境化学研究需要回答的关键科学问题之一. 随着计算机性能的提升和各种算法与软件的发展,各种计算方法已经陆续被应用于污染物环境界面行为的认识和探索中,并在环境界面化学机制探索中取得了重要进展. 尽管已有学者使用方法各异的计算手段对典型污染物的各种环境界面行为开展了广泛研究,但仍有一些问题需要进一步解决. 总体而言,污染物环境界面行为的计算模拟研究面临的核心问题是体系的环境真实性. 其一,环境是典型的多介质多界面的复杂体系,即便是单一环境介质也并非真正意义的均相体系. 譬如土壤就由土壤固体、土壤溶液和孔隙空气三相组成,而土壤固体又可分为有机固体和无机固体. 虽然计算模拟一般均采用简化的物理模型建立其数学模型,但是就如同真实土壤固相表面需要考虑水的存在而不是羟基化表面一样,大气气溶胶界面反应所在的表面也不会是彻底脱水的干表面. 然而,环境界面吸附和反应的QM计算常常忽略这一实际情况,可能造成与实际脱节的计算结果. 譬如前文提到的2021年Jiang等的工作就是很好的例证[19]. 模拟气相条件石墨烯吸附全氟烷基磺酸的QM计算与水溶液中的MD分析结果是不同的,若考虑水相纳米气泡的影响则评估结论又会发生变化,而考虑了气-液-固三相的模拟体系才更接近环境真实情况. 其二,污染物均是以与其他化学品共存的方式出现在环境中,也就是说环境界面化学研究面对的是污染物混合物而非单一污染物. 进入到环境中的不同种类污染物性质各异,各种污染物与环境界面间的相互作用也可能会对彼此产生影响[51,65]. 遗憾的是,已报道的污染物界面行为模拟研究多集中于单一污染物的环境界面化学机制的探讨,对不同类型污染物混合物的环境界面过程研究仍很少见,污染混合物的复合作用机制尚有待进一步探究. 其三,现有对环境介质化学组成的认识也限制了环境界面模型的建立和污染物与其相互作用计算模拟研究的发展. 如土壤有机质本身组成复杂,依托实验解析其分子组成和结构的全貌特征尚存在困难,各种模拟成分因此应运而生[38,75]. 这样的简化和近似也在一定程度上限制了模拟结果的环境准确性. 将计算模拟方法与实验验证融为一体,互为补充,同时借助机器学习等数据驱动的规律发掘算法,可能成为解决复杂环境介质及其参与的环境界面问题化学机制的可行途径. 其四,相较于QM与MD方法,CG模型及相应的计算模拟方法发展较晚,因此目前CG模型在污染物界面行为的计算研究中仍应用较少. 其中,CG-MD模拟的应用受限于力场参数及适用性,而DPD方法则只需要获取CG珠子间的相互作用参数即可,应用更值得关注.
污染物环境界面行为的多尺度计算模拟
Multi-scale simulation for the environmental interface behavior of pollutants
-
摘要: 污染物的环境界面行为研究一直是环境科学领域的核心和热点内容之一. 借助于计算方法能够突破实验方法分析水平的限制,提供从微观到介观的多尺度上的污染物界面行为的化学机制与结构特征. 量子力学方法、全原子分子动力学模拟和基于粗粒化体系模型的分子动力学模拟及耗散粒子动力学模拟等不同分子间作用计算方法可依次从原子尺度、分子尺度到介观尺度实现污染物环境界面行为的模拟分析,提供丰富多样的污染物与环境表面相互作用信息. 本文综述了典型分子间作用计算模拟方法在污染物的环境界面生成、吸附乃至催化转化等方面的应用进展,分析了现有研究存在的问题和不足,并对未来的研究重点提出了展望.Abstract: The research on the environmental interface behavior of pollutants has always been one of the core and hot topics in the field of environmental science. With the help of computational methods, it can break through the limitations of the analytical level of experimental methods, and to provide valuable information on the chemical mechanism and structural characteristics of pollutant interface behaviors on multi-scales from microscopic to mesoscopic. Different intermolecular interaction calculation methods, such as quantum mechanical method, all-atom molecular dynamics simulation, and dissipative particle dynamics simulation and molecular dynamics simulation based on coarse-grained system models, can realize the simulation analysis of pollutant-environment interface behavior from atomic scale, molecular scale to mesoscopic scale in turn, and provide diverse information on the interaction mechanisms of pollutants with environmental surfaces. In this paper, the application of different typical intermolecular interaction computational simulation methods in the interfacial generation, adsorption and even catalytic conversion of environmental pollutants was reviewed. The problems and limitations of the existing application research of computational methods were analyzed, and the future research focus is put forward.
-
-
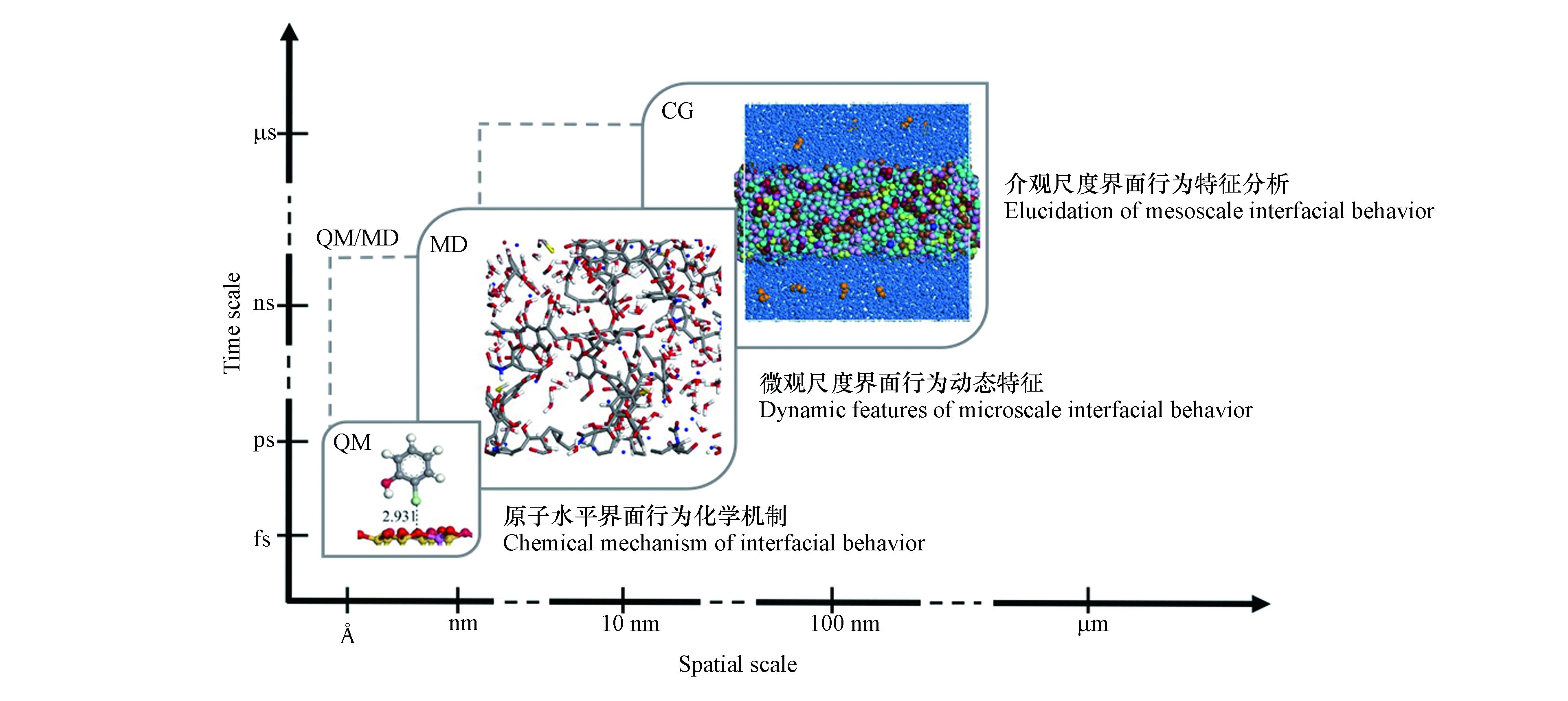
图 1 典型计算模拟方法对应的时间和空间尺度及所适应的界面问题类型
Figure 1. The time and space scales corresponding to the different computational simulation methods and the types of interfacial issues they are adapted to
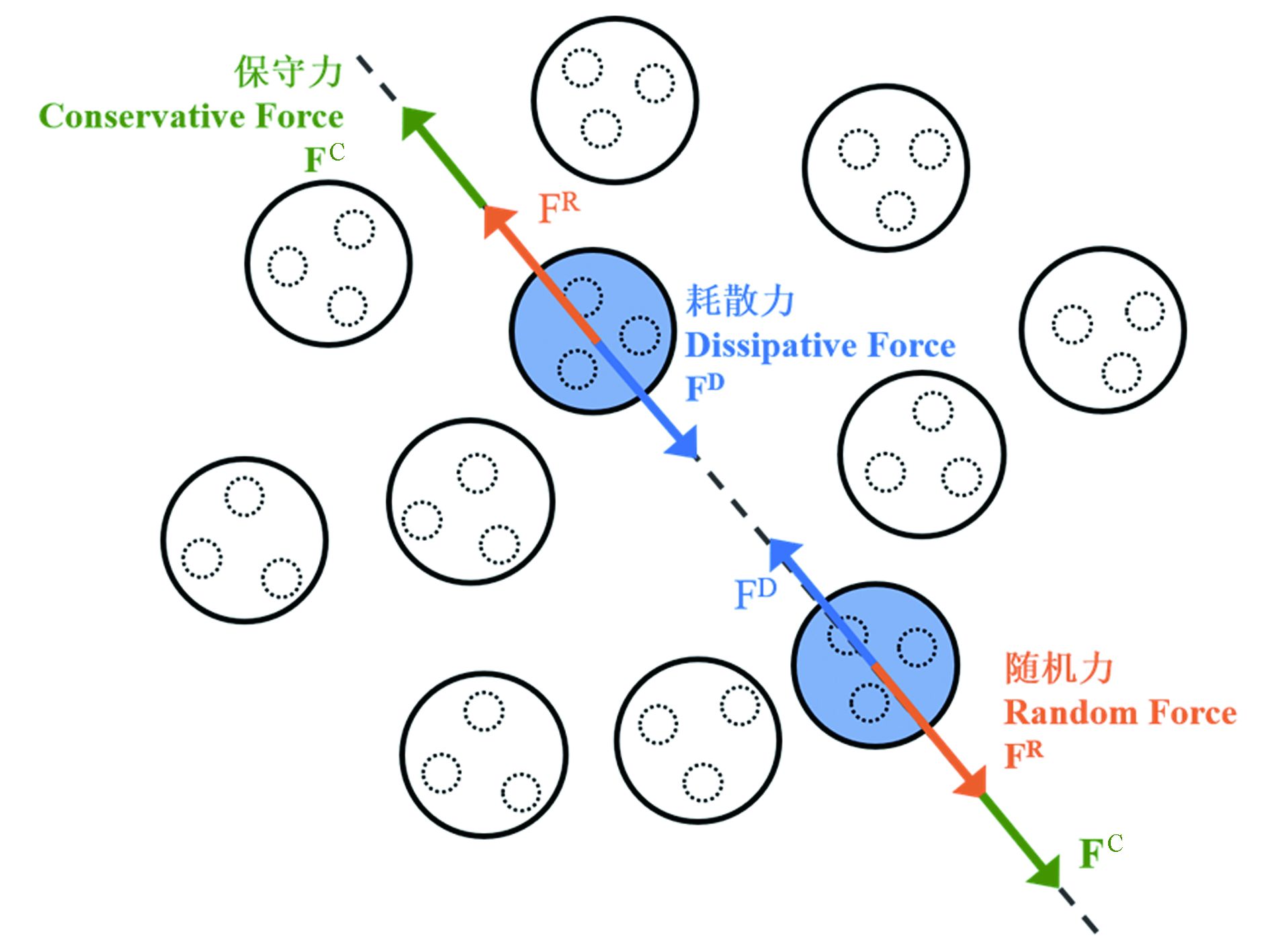
图 2 耗散粒子动力学模拟中的三种相互作用力示意图
Figure 2. Sketch of the three interaction forces in dissipative particle dynamics simulations
表 1 常用分子力场及其适用情况
Table 1. Typical molecular force fields and their applications
力场
Force field应用
ApplicationsAMBER(Assisted Model Building and Energy Refinement) 蛋白质、核酸等生物分子体系 CFF(Consistent Force Field) 有机分子、高分子及生物大分子等 CVFF(Consistent Valence Force Field) 氨基酸及水等 CHARMM(Chemistry at Harvard Molecular Mechanics) 生物大分子体系及无机材料等小分子体系 UFF(Universal Force Field) 涵盖整个周期表,包括含阳离子体系 COMPASS(Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies) 金属、金属离子及金属氧化物 MMX 有机小分子 MMFF(Merck Molecular Force Field) 有机分子 ff19SB 蛋白质体系 OPLS(Optimized Potential for Liquid Simulations) 多肽、核酸、有机溶剂等液体体系 lipid21 磷脂体系 OL21 核酸体系 GAFF(General AMBER Force Field) 有机分子 ECEPP(Empirical Conformational Energy Program for Peptides) 多肽和蛋白质分子体系 GROMOS(Groningen Molecular Simulation) 蛋白质、核酸和糖的水溶剂及极性溶液 IFF(Interface Force Field) 金属、矿物和聚合物材料界面  下载: 导出CSV
下载: 导出CSV
-
[1] 阮挺, 江桂斌. 发现新型环境有机污染物的基本理论与方法 [J]. 中国科学院院刊, 2020, 35(11): 1328-1336. doi: 10.16418/j.issn.1000-3045.20200915004 RUAN T, JIANG G B. Basic theory and analytical methodology for identification of novel environmental organic pollutants [J]. Bulletin of Chinese Academy of Sciences, 2020, 35(11): 1328-1336(in Chinese). doi: 10.16418/j.issn.1000-3045.20200915004
[2] 张佩萱, 高丽荣, 宋世杰, 等. 环境中短链和中链氯化石蜡的来源、污染特征及环境行为研究进展 [J]. 环境化学, 2021, 40(2): 371-383. doi: 10.7524/j.issn.0254-6108.2020101603 ZHANG P X, GAO L R, SONG S J, et al. Chlorinated paraffins in the environment: A review on their sources, levels and fate [J]. Environmental Chemistry, 2021, 40(2): 371-383(in Chinese). doi: 10.7524/j.issn.0254-6108.2020101603
[3] 林泳峰, 阮挺, 江桂斌. 新型全氟和多氟烷基化合物的分析、行为与效应研究进展 [J]. 科学通报, 2017, 62(24): 2724-2733. doi: 10.1360/N972017-00223 LIN Y F, RUAN T, JIANG G B. Progress on analytical methods and environmental behavior of emerging per-and polyfluoroalkyl substances [J]. Chinese Science Bulletin, 2017, 62(24): 2724-2733(in Chinese). doi: 10.1360/N972017-00223
[4] DAUGHTON C G, TERNES T A. Pharmaceuticals and personal care products in the environment: Agents of subtle change?[J]. Environmental Health Perspectives, 1999, 107(Suppl 6): 907-938. [5] MANZETTI S, van der SPOEL E R, van der SPOEL D. Chemical properties, environmental fate, and degradation of seven classes of pollutants [J]. Chemical Research in Toxicology, 2014, 27(5): 713-737. doi: 10.1021/tx500014w [6] ZHAO S, WANG J H, FENG S J, et al. Effects of ecohydrological interfaces on migrations and transformations of pollutants: A critical review [J]. Science of the Total Environment, 2022, 804: 150140. doi: 10.1016/j.scitotenv.2021.150140 [7] 曲久辉, 贺泓, 刘会娟. 典型环境微界面及其对污染物环境行为的影响 [J]. 环境科学学报, 2009, 29(1): 2-10. doi: 10.3321/j.issn:0253-2468.2009.01.002 QU J H, HE H, LIU H J. Typical environmental micro-interfaces and its effect on environmental behaviors of pollutants [J]. Acta Scientiae Circumstantiae, 2009, 29(1): 2-10(in Chinese). doi: 10.3321/j.issn:0253-2468.2009.01.002
[8] 张爱茜, 刘景富, 景传勇, 等. 我国环境化学研究新进展 [J]. 化学通报, 2014, 77(7): 654-659. doi: 10.14159/j.cnki.0441-3776.2014.07.005 ZHANG A Q, LIU J F, JING C Y, et al. Latest progresses in environmental chemistry in China [J]. Chemistry, 2014, 77(7): 654-659(in Chinese). doi: 10.14159/j.cnki.0441-3776.2014.07.005
[9] DAUGHTON C G. “Emerging” chemicals as pollutants in the environment: A 21st century perspective [J]. Renewable Resources Journal, 2005, 23(4): 6-23. [10] 李院霞, 史雪倩, 李红, 等. 同步辐射X射线荧光和吸收谱技术在环境汞污染研究中的应用进展 [J]. 生态毒理学报, 2018, 13(6): 30-38. LI Y X, SHI X Q, LI H, et al. Review of studies using synchrotron radiation X-ray fluorescence and X-ray absorption spectrum techniques on environmental Hg pollution [J]. Asian Journal of Ecotoxicology, 2018, 13(6): 30-38(in Chinese).
[11] 陈景文, 王中钰, 傅志强. 环境计算化学与毒理学[M]. 北京: 科学出版社, 2018. CHEN J W, WANG Z Y, FU Z Q. Environmental computational chemistry and toxicology [M]. Beijing: Science Press, 2018(in Chinese).
[12] TOMASI J, MENNUCCI B, CAMMI R. Quantum mechanical continuum solvation models [J]. Chemical Reviews, 2005, 36(42): 2999-3093. [13] ALLEN M P, TILDESLEY D J. Computer Simulation of Liquids[M]. Second edition. Oxford, United Kingdom: Oxford University Press, 2017. [14] OTTO D P, DE VILLIERS M M. Coarse-grained molecular dynamics (CG-MD) simulation of the encapsulation of dexamethasone in PSS/PDDA layer-by-layer assembled polyelectrolyte nanocapsules [J]. AAPS PharmSciTech, 2020, 21(8): 292. doi: 10.1208/s12249-020-01843-5 [15] LIU M B, LIU G R, ZHOU L W, et al. Dissipative Particle Dynamics (DPD): An overview and recent developments [J]. Archives of Computational Methods in Engineering, 2015, 22(4): 529-556. doi: 10.1007/s11831-014-9124-x [16] ORSI M. Molecular dynamics simulation of humic substances [J]. Chemical and Biological Technologies in Agriculture, 2014, 1: 10. doi: 10.1186/s40538-014-0010-4 [17] TRATNYEK P G, BYLASKA E J, WEBER E J. In silico environmental chemical science: Properties and processes from statistical and computational modelling [J]. Environmental Science. Processes & Impacts, 2017, 19(3): 188-202. [18] FENG H R, LIN Y, SUN Y Z, et al. In silico approach to investigating the adsorption mechanisms of short chain perfluorinated sulfonic acids and perfluorooctane sulfonic acid on hydrated hematite surface [J]. Water Research, 2017, 114: 144-150. doi: 10.1016/j.watres.2017.02.024 [19] JIANG X Z, WANG W, YU G, et al. Contribution of nanobubbles for PFAS adsorption on graphene and OH- and NH2-functionalized graphene: Comparing simulations with experimental results [J]. Environmental Science & Technology, 2021, 55(19): 13254-13263. [20] 林梦海. 量子化学简明教程[M]. 北京: 化学工业出版社, 2005. LIN M H. Concise course in quantum chemistry [M]. Beijing: Chemical Industry Press, 2005(in Chinese).
[21] van der SPOEL D, MANZETTI S, ZHANG H Y, et al. Prediction of partition coefficients of environmental toxins using computational chemistry methods [J]. ACS Omega, 2019, 4(9): 13772-13781. doi: 10.1021/acsomega.9b01277 [22] FU W J, XIA G J, ZHANG Y X, et al. Using general computational chemistry strategy to unravel the reactivity of emerging pollutants: An example of sulfonamide chlorination [J]. Water Research, 2021, 202: 117391. doi: 10.1016/j.watres.2021.117391 [23] MILMAN V, WINKLER B, WHITE J A, et al. Electronic structure, properties, and phase stability of inorganic crystals: A pseudopotential plane-wave study [J]. International Journal of Quantum Chemistry, 2000, 77(5): 895-910. doi: 10.1002/(SICI)1097-461X(2000)77:5<895::AID-QUA10>3.0.CO;2-C [24] MAZUREK A H, SZELESZCZUK Ł, PISKLAK D M. Periodic DFT calculations-review of applications in the pharmaceutical sciences [J]. Pharmaceutics, 2020, 12(5): 415. doi: 10.3390/pharmaceutics12050415 [25] HOHENBERG P, KOHN W. Inhomogeneous electron gas [J]. Physical Review, 1964, 136(3B): B864-B871. doi: 10.1103/PhysRev.136.B864 [26] KOHN W, BECKE A D, PARR R G. Density functional theory of electronic structure [J]. The Journal of Physical Chemistry, 1996, 100(31): 12974-12980. doi: 10.1021/jp960669l [27] RAMACHANDRAN K I, DEEPA G, NAMBOORI K. Computational chemistry and molecular modeling[M]. Berlin: Springer Berlin Heidelberg, 2008 [28] RAPAPORT D. The art of molecular dynamics simulation [M]. 2nd ed. Cambridge: Cambridge University Press, 2004 [29] 陈正隆, 徐为人, 汤立达. 分子模拟的理论与实践[M]. 北京: 化学工业出版社, 2007. CHEN Z L, XU W R, TANG L D. Theory and practice of molecular simulation [M]. Beijing: Chemical Industry Press, 2007(in Chinese).
[30] KLEIN M L, SHINODA W. Large-scale molecular dynamics simulations of self-assembling systems [J]. Science, 2008, 321(5890): 798-800. doi: 10.1126/science.1157834 [31] JOSHI S Y, DESHMUKH S A. A review of advancements in coarse-grained molecular dynamics simulations [J]. Molecular Simulation, 2021, 47(10/11): 786-803. [32] ALESSANDRI R, GRÜNEWALD F, MARRINK S J. The martini model in materials science [J]. Advanced Materials, 2021, 33(24): 2008635. doi: 10.1002/adma.202008635 [33] HOOGERBRUGGE P J, KOELMAN J M V A. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics [J]. Europhysics Letters, 1992, 19(3): 155-160. doi: 10.1209/0295-5075/19/3/001 [34] KONG Y, MANKE C W, MADDEN W G, et al. Simulation of a confined polymer in solution using the dissipative particle dynamics method [J]. International Journal of Thermophysics, 1994, 15(6): 1093-1101. doi: 10.1007/BF01458818 [35] ESPAÑOL P. Hydrodynamics from dissipative particle dynamics [J]. Physical Review E, 1995, 52(2): 1734-1742. doi: 10.1103/PhysRevE.52.1734 [36] GROOT R D, WARREN P B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation [J]. The Journal of Chemical Physics, 1997, 107(11): 4423-4435. doi: 10.1063/1.474784 [37] GUO F Y, XU J C, FEIN J B, et al. Crystal face-dependent methylmercury adsorption onto mackinawite (FeS) nanocrystals: A DFT-D3 study [J]. Chemical Engineering Journal, 2021, 420: 127594. doi: 10.1016/j.cej.2020.127594 [38] AHMED A A, THIELE-BRUHN S, LEINWEBER P, et al. Towards a molecular level understanding of the sulfanilamide-soil organic matter-interaction [J]. Science of the Total Environment, 2016, 559: 347-355. doi: 10.1016/j.scitotenv.2016.03.136 [39] LI J, WU Y L, BAI H H, et al. Highly efficient adsorption and mechanism of alkylphenols on magnetic reduced graphene oxide [J]. Chemosphere, 2021, 283: 131232. doi: 10.1016/j.chemosphere.2021.131232 [40] ZOU M Y, ZHANG J D, CHEN J W, et al. Simulating adsorption of organic pollutants on finite (8, 0) single-walled carbon nanotubes in water [J]. Environmental Science & Technology, 2012, 46(16): 8887-8894. [41] MOSALLANEJAD S, DLUGOGORSKI B Z, KENNEDY E M, et al. Formation of PCDD/fs in oxidation of 2-chlorophenol on neat silica surface [J]. Environmental Science & Technology, 2016, 50(3): 1412-1418. [42] PAN W X, CHANG J M, LIU X, et al. Interfacial formation of environmentally persistent free radicals—A theoretical investigation on pentachlorophenol activation on montmorillonite in PM2.5 [J]. Ecotoxicology and Environmental Safety, 2019, 169: 623-630. doi: 10.1016/j.ecoenv.2018.11.041 [43] ASSAF N W, ALTARAWNEH M, OLUWOYE I, et al. Formation of environmentally persistent free radicals on α-Al2O3 [J]. Environmental Science & Technology, 2016, 50(20): 11094-11102. [44] LOMNICKI S, TRUONG H, VEJERANO E, et al. Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter [J]. Environmental Science & Technology, 2008, 42(13): 4982-4988. [45] SAKR N I, KIZILKAYA O, CARLSON S F, et al. Formation of environmentally persistent free radicals (EPFRs) on the phenol-dosed α-Fe2O3(0001) surface [J]. The Journal of Physical Chemistry C, Nanomaterials and Interfaces, 2021, 125(40): 21882-21890. doi: 10.1021/acs.jpcc.1c04298 [46] AHMED O H, ALTARAWNEH M, AL-HARAHSHEH M, et al. Formation of phenoxy-type environmental persistent free radicals (EPFRs) from dissociative adsorption of phenol on Cu/Fe and their partial oxides [J]. Chemosphere, 2020, 240: 124921. doi: 10.1016/j.chemosphere.2019.124921 [47] PAN W X, CHANG J M, HE S M, et al. Major influence of hydroxyl and nitrate radicals on air pollution by environmentally persistent free radicals [J]. Environmental Chemistry Letters, 2021, 19(6): 4455-4461. doi: 10.1007/s10311-021-01278-9 [48] LIU C, MIN Y, ZHANG A Y, et al. Electrochemical treatment of phenol-containing wastewater by facet-tailored TiO2: Efficiency, characteristics and mechanisms [J]. Water Research, 2019, 165: 114980. doi: 10.1016/j.watres.2019.114980 [49] HUANG M J, HAN Y, XIANG W, et al. In situ-formed phenoxyl radical on the CuO surface triggers efficient persulfate activation for phenol degradation [J]. Environmental Science & Technology, 2021, 55(22): 15361-15370. [50] GAO M, HE G, ZHANG W, et al. Reaction pathways of the selective catalytic reduction of NO with NH3 on the α-Fe2O3 (012) surface: A combined experimental and DFT study [J]. Environmental Science & Technology, 2021, 55(16): 10967-10974. [51] SUI H, LI L, ZHU X Z, et al. Modeling the adsorption of PAH mixture in silica nanopores by molecular dynamic simulation combined with machine learning [J]. Chemosphere, 2016, 144: 1950-1959. doi: 10.1016/j.chemosphere.2015.10.053 [52] ARISTILDE L, MARICHAL C, MIÉHÉ-BRENDLÉ J, et al. Interactions of oxytetracycline with a smectite clay: A spectroscopic study with molecular simulations [J]. Environmental Science & Technology, 2010, 44(20): 7839-7845. [53] KERISIT S, LIU C X. Molecular dynamics simulations of uranyl and uranyl carbonate adsorption at aluminosilicate surfaces [J]. Environmental Science & Technology, 2014, 48(7): 3899-3907. [54] WILLEMSEN J A R, MYNENI S C B, BOURG I C. Molecular dynamics simulations of the adsorption of phthalate esters on smectite clay surfaces [J]. The Journal of Physical Chemistry C, 2019, 123(22): 13624-13636. doi: 10.1021/acs.jpcc.9b01864 [55] ZHU X Z, CHEN D Y, WU G Z. Molecular dynamic simulation of asphaltene co-aggregation with humic acid during oil spill [J]. Chemosphere, 2015, 138: 412-421. doi: 10.1016/j.chemosphere.2015.06.074 [56] PETROV D, TUNEGA D, GERZABEK M H, et al. Molecular modelling of sorption processes of a range of diverse small organic molecules in Leonardite humic acid [J]. European Journal of Soil Science, 2020, 71(5): 831-844. [57] VIALYKH E A, SALAHUB D R, ACHARI G, et al. Emergent functional behaviour of humic substances perceived as complex labile aggregates of small organic molecules and oligomers [J]. Environmental Chemistry, 2019, 16(7): 505. doi: 10.1071/EN19095 [58] TUNEGA D, GERZABEK M H, HABERHAUER G, et al. Adsorption process of polar and nonpolar compounds in a nanopore model of humic substances [J]. European Journal of Soil Science, 2020, 71(5): 845-855. [59] ZHAO N, JU F, PAN H, et al. Molecular dynamics simulation of the interaction of water and humic acid in the adsorption of polycyclic aromatic hydrocarbons [J]. Environmental Science and Pollution Research International, 2020, 27(20): 25754-25765. doi: 10.1007/s11356-020-09018-2 [60] 林文强, 徐斌, 陈亮, 等. 双酚A在氧化石墨烯表面吸附的分子动力学模拟 [J]. 物理学报, 2016, 65(13): 86-92. doi: 10.7498/aps.65.133102 LIN W Q, XU B, CHEN L, et al. Molecular dynamics simulations of the adsorption of bisphenol A on graphene oxide [J]. Acta Physica Sinica, 2016, 65(13): 86-92(in Chinese). doi: 10.7498/aps.65.133102
[61] WANG Y, COMER J, CHEN Z F, et al. Exploring adsorption of neutral aromatic pollutants onto graphene nanomaterials via molecular dynamics simulations and theoretical linear solvation energy relationships [J]. Environmental Science:Nano, 2018, 5: 2117-2128. doi: 10.1039/C8EN00575C [62] TANG H, ZHAO Y, SHAN S J, et al. Theoretical insight into the adsorption of aromatic compounds on graphene oxide [J]. Environmental Science:Nano, 2018, 5(10): 2357-2367. doi: 10.1039/C8EN00384J [63] TANG H, ZHAO Y, YANG X N, et al. Understanding the pH-dependent adsorption of ionizable compounds on graphene oxide using molecular dynamics simulations [J]. Environmental Science:Nano, 2017, 4(10): 1935-1943. doi: 10.1039/C7EN00585G [64] TANG H, ZHAO Y, SHAN S J, et al. Wrinkle- and edge-adsorption of aromatic compounds on graphene oxide as revealed by atomic force microscopy, molecular dynamics simulation, and density functional theory [J]. Environmental Science & Technology, 2018, 52(14): 7689-7697. [65] 赵超锋, 金佳人, 霍英忠, 等. 氧化石墨烯吸附水体中酚类有机污染物的分子动力学模拟 [J]. 无机材料学报, 2020, 35(3): 277-285. doi: 10.15541/jim20190377 ZHAO C F, JIN J R, HUO Y Z, et al. Adsorption of phenolic organic pollutants on graphene oxide: Molecular dynamics study [J]. Journal of Inorganic Materials, 2020, 35(3): 277-285(in Chinese). doi: 10.15541/jim20190377
[66] SCHWARZENBACH R P, ESCHER B I, FENNER K, et al. The challenge of micropollutants in aquatic systems [J]. Science, 2006, 313(5790): 1072-1077. doi: 10.1126/science.1127291 [67] GEYER R, JAMBECK J R, LAW K L. Production, use, and fate of all plastics ever made [J]. Science Advances, 2017, 3(7): e1700782. doi: 10.1126/sciadv.1700782 [68] RAMALHO J P P, DORDIO A V, CARVALHO A J P. The fate of three common plastic nanoparticles in water: A molecular dynamics study [J]. Journal of Molecular Structure, 2022, 1249: 131520. doi: 10.1016/j.molstruc.2021.131520 [69] GUO X, LIU Y, WANG J L. Equilibrium, kinetics and molecular dynamic modeling of Sr2+ sorption onto microplastics [J]. Journal of Hazardous Materials, 2020, 400: 123324. doi: 10.1016/j.jhazmat.2020.123324 [70] LI H, WANG F H, LI J N, et al. Adsorption of three pesticides on polyethylene microplastics in aqueous solutions: Kinetics, isotherms, thermodynamics, and molecular dynamics simulation [J]. Chemosphere, 2021, 264: 128556. doi: 10.1016/j.chemosphere.2020.128556 [71] CHEN Y J, LI J N, WANG F H, et al. Adsorption of tetracyclines onto polyethylene microplastics: A combined study of experiment and molecular dynamics simulation [J]. Chemosphere, 2021, 265: 129133. doi: 10.1016/j.chemosphere.2020.129133 [72] MOORE M N. Do nanoparticles present ecotoxicological risks for the health of the aquatic environment? [J]. Environment International, 2006, 32(8): 967-976. doi: 10.1016/j.envint.2006.06.014 [73] LADO TOURIÑO I, NARANJO A C, NEGRI V, et al. Coarse-grained molecular dynamics simulation of water diffusion in the presence of carbon nanotubes [J]. Journal of Molecular Graphics and Modelling, 2015, 62: 69-73. doi: 10.1016/j.jmgm.2015.09.009 [74] WANG Z, QUIK J T K, SONG L, et al. Dissipative particle dynamic simulation and experimental assessment of the impacts of humic substances on aqueous aggregation and dispersion of engineered nanoparticles [J]. Environmental Toxicology and Chemistry, 2018, 37(4): 1024-1031. doi: 10.1002/etc.4059 [75] DETTMANN L F, KÜHN O, AHMED A A. Coarse-grained molecular dynamics simulations of nanoplastics interacting with a hydrophobic environment in aqueous solution [J]. RSC Advances, 2021, 11(44): 27734-27744. doi: 10.1039/D1RA04439G [76] FENG H R, ZHANG H Y, CAO H M, et al. Application of a novel coarse-grained soil organic matter model in the environment [J]. Environmental Science & Technology, 2018, 52(24): 14228-14234. [77] RUIZ-MORALES Y, MULLINS O C. Coarse-grained molecular simulations to investigate asphaltenes at the oil-water interface [J]. Energy & Fuels, 2015, 29(3): 1597-1609. [78] DING N, CHEN X F, WU C M L. Interactions between polybrominated diphenyl ethers and graphene surface: A DFT and MD investigation [J]. Environmental Science:Nano, 2014, 1(1): 55-63. doi: 10.1039/C3EN00037K [79] CORTÉS-ARRIAGADA D, TORO-LABBÉ A. A theoretical investigation of the removal of methylated arsenic pollutants with silicon doped graphene [J]. RSC Advances, 2016, 6(34): 28500-28511. doi: 10.1039/C6RA03813A [80] ZHANG C, LIU X D, LU X C, et al. Understanding the heterogeneous nucleation of heavy metal phyllosilicates on clay edges with first-principles molecular dynamics [J]. Environmental Science & Technology, 2019, 53(23): 13704-13712. -
 点击查看大图
点击查看大图
计量
- 文章访问数: 7401
- HTML全文浏览数: 7401
- PDF下载数: 225
- 施引文献: 0
























