

-
活性污泥法是目前世界上应用最成功的生物处理技术[1],在水环境保护和公共卫生保障中发挥着重要作用。活性污泥法以细菌群落为主体,含有的细菌种类繁多[2],但又不是细菌细胞的简单聚集,而是不同细菌相互合作形成的具有复杂物理结构的絮体菌胶团[3],从而保证了降解菌在处理系统中的持留和碳、氮、磷等污染物的高效去除[4]。值得注意的是,上述组成复杂的活性污泥微生物群落是由进水及环境中分散的细菌聚集、增殖和相互作用形成的[5-6],解析该形成过程中细菌群落的演替及其对运行参数或环境压力的响应对于深入理解活性污泥降解污染物的微生态机制具有重要指导意义。
李培睿等[7]将活性污泥形成的宏观过程分为4个阶段:细菌增殖阶段、絮状体形成阶段、絮状体聚合阶段和凝絮体形成阶段,其中初期的细菌增殖阶段决定了活性污泥最终的细菌群落组成,对活性污泥微生物群落构建至关重要。李培睿等[7]使用光学和扫描电子显微镜观察发现,活性污泥形成初期以球状游离细菌为主,后期杆菌和丝状菌大量增殖并逐渐形成菌胶团,表明活性污泥初期增殖阶段有明显的微生物群落演替。在群落构建的影响因素方面, GAO等[8]发现初始菌种比例可以改变细菌之间的相互作用关系,从而影响细菌群落的结构和功能。RATZKE等[9]发现高营养条件会促进细菌间的竞争,导致群落多样性降低,进而降低最终群落的稳定性。除了接种比例和营养水平外,环境压力也是影响活性污泥细菌群落构建的一个关键因素[6]。随着抗生素的广泛使用,环境水体中抗生素检出频率和浓度均逐渐有所增加[10-12],在畜禽养殖、医院和制药等污水中抗生素质量浓度甚至可达到mg·L−1[13-17],使得抗生素类药物成为一种潜在影响活性污泥形成过程的环境压力。抗生素如四环素和诺氟沙星等在低浓度时具有信号分子的作用,可以促进细菌的移动和聚集[18]。低浓度四环素还可选择性富集硝化细菌和聚磷菌[19],进而有可能促进活性污泥的形成。而高浓度抗生素具有抑菌和杀菌作用,可显著抑制硝化菌和反硝化菌增殖[19],并可导致活性污泥解体[20]。因此,抗生素在不同的浓度水平下可能会对活性污泥微生物群落的构建过程产生不同的影响,但相关研究还未见报道。
本研究选择了中国使用量大的第2代四环素米诺环素作为模型化合物[21],设置无抗生素暴露对照组和5个抗生素浓度梯度,通过宏观观测和微生物群落解析,考察了不同抗生素暴露压力下活性污泥初期增殖阶段细菌群落的构建过程及其微生态机制。本研究结果可为含抗生素废水的高效生化处理提供参考。
-
米诺环素盐酸盐(纯度≥98%)购自上海麦克林生化科技有限公司,用纯水配成10 000 mg·L−1储备液;活性污泥微生物增殖使用的培养液为LB肉汤培养基,购自青岛高科园海博生物技术有限公司;活性污泥形成过程的生物样品采用0.22 μm聚碳酸酯滤膜抽滤收集,滤膜购自默克密理博有限公司(德国),过滤后水样用于化学需氧量(COD)测定;其余化学试剂均为分析纯,购自天津市科密欧化学试剂有限公司。用于制备污泥增殖实验种泥的活性污泥取自石家庄某市政污水处理厂的曝气池,污泥质量浓度为7 780 mg·L−1。
-
将15 mL市政污水厂取的活性污泥加入到6 L自来水中,快速搅拌30 min以充分打散污泥,将打散后的溶液(污泥质量浓度约20 mg·L−1)均匀分装到48个150 mL锥形瓶中,每瓶100 mL。从每个锥形瓶中取出8 mL混合液作为初始活性污泥样品(第0天),代表本研究活性污泥细菌群落构建的种泥,然后加入米诺环素储备液以达到预期抗生素暴露质量浓度和LB营养液使溶液COD质量浓度达到约400 mg·L−1,不足体积使用超纯水补齐。使用组培封口膜将锥形瓶封口后放入恒温(25 ℃,转速150 r·min−1)振荡培养箱开始实验。设置无抗生素压力的对照组和米诺环素质量浓度分别为0.001、0.01、0.1、1和10 mg·L−1的实验组,共6组,每组8个平行。
实验过程中,每2 d从锥形瓶中取样1次,取样前充分混匀,取样体积为8 mL,取样后加入抗生素储备液以维持抗生素质量浓度,加入新鲜LB营养液保证混合液中重新补充约400 mg·L−1 COD以支持污泥的增殖,并加水保证反应体系总体积维持在100 mL,实验周期20 d。
-
活性污泥增殖过程的生物量(混合液悬浮固体浓度,MLSS)和溶解性COD参照国标法测定[22]。用相机拍照记录活性污泥的宏观形态变化。对照组和实验组分别在第0、4、10、20天取4 mL混合液,通过0.22 μm滤膜过滤收集生物样,用于DNA提取和细菌群落分析。DNA提取使用FastDNA©SPIN Kit for Soil (Qbiogene,USA)试剂盒,提取后DNA使用515F (GTGCCAGCMGCCGCGG)和907R (CCGTCAATTCMTTTRAGTTT)引物扩增细菌16S rRNA基因[23],PCR扩增产物送至上海美吉生物医药科技有限公司进行Miseq测序。测序数据存储于NCBI SRA,链接号为PRJNA783472。测序数据质控、OTU(operational taxonomic units)聚类及细菌菌属注释通过美吉生物云平台(https://cloud.majorbio.com/)完成。
-
使用香农指数(Shannon Diversity Index)表征细菌群落的α多样性,使用非度量多维尺度分析(NMDS)揭示活性污泥初期增殖阶段细菌群落结构的变化(β多样性),两分析同样在美吉生物云平台完成。使用Origin 8.0进行配对t检验以分析组间差异的显著性。为了评估不同抗生素压力下活性污泥细菌群落的紧密程度,基于16S rRNA基因的Miseq测序数据,通过在线平台(http://ieg2.ou.edu/MENA)构建了由原核微生物群落成员组成的分子生态网络,使用Gephi 0.92软件进行网络可视化。
-
图1反映了在不同米诺环素压力下活性污泥的宏观增殖过程。第0天,对照组和实验组摇瓶内溶液均透明,无污泥产生。0.001~0.1 mg·L−1实验组与对照组的污泥宏观形态变化相似,随着培养时间延长,逐渐生成黄褐色颗粒状絮体。1 mg·L−1实验组在第4天同样产生了少量颗粒状污泥絮体,但体系中游离细菌较多,导致混合液略显浑浊,继续培养后第10天和第20天照片显示游离细菌减少,颗粒状污泥絮体增多,上清液逐渐澄清,第20天的污泥宏观形态与低质量浓度抗生素暴露组和对照组相似。10 mg·L−1实验组污泥形态与其他实验组明显不同,在20 d内未形成污泥絮体,且混合液整体呈现浑浊状态,长时间静置底部有少量分散的菌斑沉淀(图1)。这表明米诺环素在该浓度下可严重抑制活性污泥絮体的形成。
-
通过测定实验过程的污泥和溶解性耗氧有机物(以COD计)的质量浓度来定量表征污泥的生物量和代谢活性变化。如图2(a)所示,污泥初始增殖阶段的生物量较低且呈聚集态增殖。采样不均一及测定偏差大导致MLSS的数值偏差较大,但总体上对照组和实验组污泥质量浓度的变化趋势相似。实验开始后MLSS快速增加,在第10~14天进入平台期,6个体系的最终污泥质量浓度均在400~500 mg·L−1,组间生物量不存在显著差异(P>0.05)。初始增殖阶段,由于污泥量低,无法完全代谢加入的营养基质,导致溶解性耗氧有机物(以COD计)的累积,各反应组的溶解性耗氧有机物(以COD计)均在第8天达到最大值,之后随着污泥量增加,对营养基质利用率增加,对照组和0.001~1 mg·L−1实验组的溶解性耗氧有机物(以COD计)逐渐下降,并在第12天重新达到平衡直至实验结束(图2(b))。10 mg·L−1实验组的溶解性耗氧有机物(以COD计)也表现出类似的变化趋势,不过最终稳定的COD值(约200 mg·L−1)显著高于1 mg·L−1(约130 mg·L−1)和其他实验组(约70 mg·L−1)(图2(b)),表明10 mg·L−1实验组最终形成的细菌群落具有较弱的有机物降解能力。同时,污泥质量浓度和溶解性耗氧有机物(以COD计)的变化趋势(图2)及污泥宏观形貌变化结果(图1)也表明,本研究选择的20 d实验周期内活性污泥细菌群落增殖已达到设置实验条件(营养水平、温度和采样模式等)下的稳态,可以开展后续的微生态解析。
-
细菌群落是活性污泥的核心,解析其结构、关键物种和作用机制是提高工艺运行效果的重要依据[24]。使用16S rRNA基因扩增子测序分析了不同米诺环素压力下活性污泥形成过程的细菌群落动态变化,192个污泥样本共得到近600万条高质量原核微生物16S rRNA序列,这些序列被划分为4 795个OTUs。在α多样性方面,种泥细菌多样性较高,香农多样性指数为5.6。对照组和0.001~0.1 mg·L−1实验组在实验的第4天香农指数快速降低至约3.5,之后基本保持不变(图3(a)),细菌群落多样性的降低应该主要与实验使用的营养液成分相对单一且浓度较高有关[9,25]。1 mg·L−1实验组在第4天后多样性继续下降,在第20天降低至2.8,表明1 mg·L−1米诺环素已经可以影响活性污泥的细菌多样性。10 mg·L−1米诺环素降低污泥细菌种群多样性的作用更加显著,第4天香农指数即降低至1.6,实验结束时更是降低至0.8(图3(a))。TIAN等[26-27]在研究第1代四环素土霉素对好氧活性污泥和中温厌氧消化的影响时,同样发现随着土霉素质量浓度增加好氧污泥和厌氧消化污泥的细菌群落多样性均显著降低,表明高质量浓度四环类抗生素暴露会使少量细菌获得绝对的生存优势,导致细菌群落的高度单一性。本研究不同组别的细菌群落组成差异(β多样性)如图3(b)所示。对照组和0.001~0.1 mg·L−1实验组细菌群落演替规律高度一致,表明0.1 mg·L−1及以下质量浓度的米诺环素不会影响活性污泥细菌群落的形成;1 mg·L−1米诺环素暴露对活性污泥增殖初期(第4天)细菌群落的影响较大,但是随着实验进行,其群落结构与低质量浓度暴露组或对照组的相似度逐渐增加;10 mg·L−1米诺环素会导致活性污泥细菌群落演替完全偏离无抗生素压力下细菌群落的演替轨迹,表明该浓度水平下米诺环素会严重干扰活性污泥的初期增殖过程。
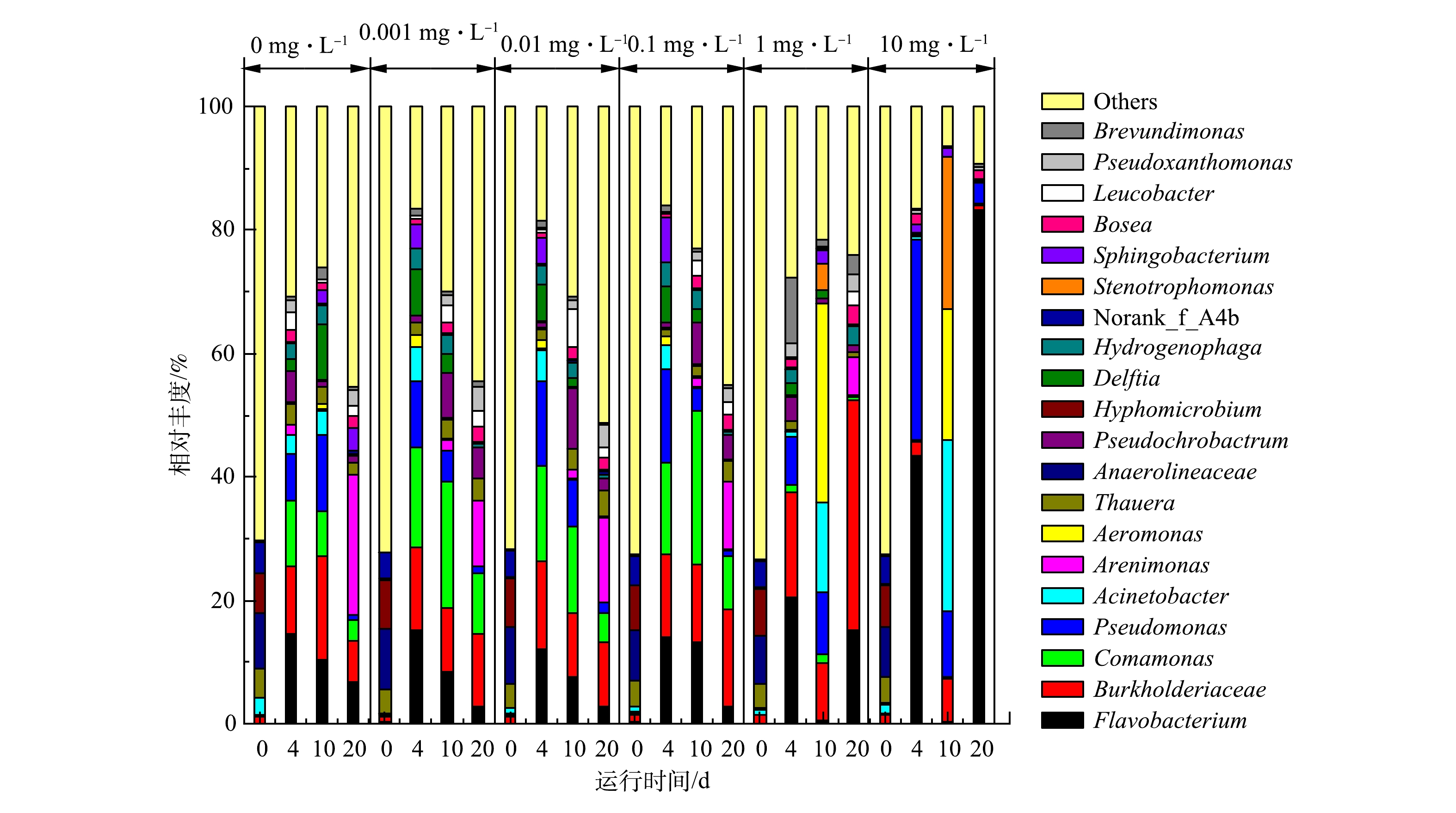
细菌群落组成方面,种泥细菌群落主要由Proteobacteria(35%)、Chloroflexi(26%)、Actinobacteria(10%)、Planctomycetes(7%)和Bacteroidetes(6%)等细菌组成(图4),与常规活性污泥细菌群落组成一致[28-31]。在第20天实验结束时,对照组和0.001~1 mg·L−1实验组细菌群落主要由Proteobacteria(63%~69%)和Bacteroidetes(18%~24%)组成,且Proteobacteria仍是最主要的细菌类别,而在10 mg·L−1实验组占主导的却是Bacteroidetes(85%),其次是Proteobacteria(14%)(图4)。Proteobacteria菌门包括了异养菌(Hyphomicrobium属于Alphaproteobacteria)、反硝化菌(Comamonas和Thauera属于Betaproteobacteria)、硝化细菌(Nitrosomonas属于Betaproteobacteria)和聚磷菌(Acinetobacter属于Gammaproteobacteria)等大量活性污泥关键功能菌[24],承担着主要的污染物降解功能,通常是活性污泥中含量最高的细菌类别,丰度可达到36%~65%,而Bacteroidetes主要包含一些异养菌,在常规活性污泥中的比例较低(2.7%~15.6%)[28]。本研究中10 mg·L−1暴露组Bacteroidetes的大量增殖,表明其可能具有米诺环素耐药性,从而在高质量浓度米诺环素暴露下获得更大的生存优势,而Proteobacteria菌门细菌多为米诺环素敏感菌,在高质量浓度米诺环素压力下丰度显著降低(图4),同时造成其体系中残留COD较高(图2(b))。
为了深入解析细菌群落的功能性适应,进一步在属水平上分析了实验期间细菌群落的演替情况。种泥的细菌群落中高丰度的菌属为norank_f__Anaerolineaceae(8.61%)、Hyphomicrobium(7.30%)、norank_f__A4b(4.45%)和Thauera(4.16%)(图5)。在实验结束时(20 d),对照组和0.001~0.1 mg·L−1实验组高丰度的菌属变为Arenimonas(10.71%~22.64%)、unclassified_f__Burkholderiaceae(6.66%~16.42%)、Comamonas(3.51%~9.94%)、Flavobacterium(2.61%~6.76%)。1 mg·L−1实验组占主导的菌属变为unclassified_f__Burkholderiaceae(37.39%)、Flavobacterium(15.01%)、Arenimonas(6.30%)和Hydrogenophaga(3.12%)。其中Burkholderiaceae细菌是活性污泥中常见的有机物降解菌,通常具有反硝化功能[32],随着其成为主导细菌,活性污泥细菌群落的多样性和均匀性明显降低。10 mg·L−1实验组的细菌群落多样性和均匀性进一步降低,Flavobacterium成为绝对主导的细菌,相对丰度达到83.14%,其次为Pseudomonas(3.33%)、Bosea(1.38%)和unclassified_f__Enterobacteriaceae(1.26%)(图5)。Flavobacterium属于Bacteroidetes,是一种革兰氏阴性、兼性厌氧菌[33],具有高生长速率,可分泌单加氧酶和水解酶高效降解纤维素等复杂碳水化合物,广泛分布于活性污泥中[34]。最近,TIAN等[35]发现Flavobacterium是水环境中四环素抗性基因tetX的天然宿主,tetX编码的蛋白可化学修饰米诺环素使其失活[36],从而使得Flavobacterium获得更大的生存优势,成为高质量浓度米诺环素暴露下的主导菌属,后续研究需结合筛菌和耐药性表型及基因型检测验证该猜测。
-
为解析在不同米诺环素压力下活性污泥增殖过程中细菌间相互作用及其变化,通过筛选并保留在50%的样本中同时出现的OTU,构建了细菌物种间的互作网络(图6)。互作网络在不同抗生素暴露浓度条件下均以相同的相似阈值(0.71)构建,使不同网络的拓扑结构参数可以直接进行比较(图7)。网络图依据细菌所属门类进行着色,图7中每一个节点代表着一个OTU,节点的大小与其连接度正相关,节点之间的连线代表着物种之间存在显著的相互作用关系,红色代表负相关,蓝色代表正相关。如图6和图7所示,1 mg·L−1和10 mg·L−1暴露组中网络拥有更少的节点和边,以及更低的平均连接度,但节点之间的正相关关系比例(相关系数为1)(41%和43%)明显高于对照组(30%)和低质量浓度暴露组(29%),表明高质量浓度米诺环素虽然降低了形成的活性污泥细菌群落复杂度和稳定性,但是却增强了存活微生物之间的合作关系[37]。WANG等[38]同样发现低降水量会降低半干旱草地土壤微生物互作网络的复杂度,但却增强了网络节点间的正相关关系。HERNANDEZ等[39]在研究另一种环境压力海拔对土壤微生物群落稳定性的影响时也得到了类似的结果。
根据微生物物种之间的互作关系,他们以相互独立或联系的模块(module)出现在群落中,对复杂网络中模块的识别可以进一步揭示网络的组织结构和形成机制[40]。对照组和0.001~10 mg·L−1实验组分别得到4、3、3、2、2、3个相互分离的模块(图7)。模块内共存的类群被认为具有共同的环境偏好或是以相互促进的方式发生互作[37]。根据节点的模块内连通性和模块间连通性将节点区分为外围节点(peripherals)、连接节点(connectors)、模块中心点(module hubs)和网络中心点(network hubs),其中连接节点、模块中心点和网络中心点被当作分子生态网络中的关键物种[37]。本研究中每个网络的大多数(>77%)节点均为外围节点,仅在0.01 mg·L−1暴露组发现了一个网络中心点,属于Hyphomicrobium(表1)。Hyphomicrobium是活性污泥中常见的异养菌[41],对于甲醇等一碳有机物有较高的去除能力[42],其在0.01 mg·L−1米诺环素暴露下成为活性污泥细菌群落的超级泛化种(super generalists),对于维持整体网络及其所属独立模块的稳定都具有重要意义[43]。连接节点是与其他模块高度连接的节点,其在不同网络中的数量变化较大:对照组为10个,0.001 mg·L−1和0.01 mg·L−1暴露组分别增加至71个和51个,而在0.1~10 mg·L−1暴露组中未找到或仅找到少量连接节点(表1)。连接节点可以增强网络的复杂度,提升网络稳定性。上述结果表明低质量浓度(0.001 mg·L−1和0.01 mg·L−1)米诺环素可能会促进活性污泥增殖过程中细菌间互作,这可能与其在低质量浓度下具有信号因子作用有关[18]。模块中心点是与自己所属模块中的许多其他节点高度连接的节点,是维持模块稳定性的关键物种。对照组及0.001~10 mg·L−1实验组检出的模块中心点数分别是4、3、4、4、6和1个(表1),这些模块中心点大多属于Proteobacteria(16/22),其次是Actinobacteria(3/22)、Bacteroidetes(2/22)和Chloroflexi(1/22)。这表明Proteobacteria细菌可能是活性污泥初期增殖阶段最为重要的关键物种。同时,需要指出网络分析揭示的关键细菌并不是细菌群落中的高丰度菌属,最典型的例子既是10 mg·L−1米诺环素暴露组网络关键物种为Proteobacteria的Rheinheimera(表1),而此时Bacteroidetes是占绝对优势(85%)的细菌类别(图4)。WANG等[38]在研究降水强度对半干旱草地土壤微生物间相互作用的影响时也发现了类似的现象。
-
1)活性污泥初期增殖阶段的宏观生物量增长与米诺环素浓度无关,10 mg·L−1米诺环素会抑制活性污泥絮体结构的形成。
2)1 mg·L−1的米诺环素即可导致活性污泥初期增殖阶段细菌多样性的降低,而10 mg·L−1的米诺环素暴露对多样性的影响更加显著;米诺环素主要富集了活性污泥中Bacteroidetes的Flavobacterium菌属。
3)低质量浓度米诺环素(0.001 mg·L−1和0.01 mg·L−1)会促进活性污泥细菌群落不同模块间的互作,从而可提升群落稳定性;而高质量浓度(1 mg·L−1和10 mg·L−1)暴露会显著降低细菌互作网络的复杂度,却促进了存活细菌间的合作关系。
4)Proteobacteria细菌是活性污泥初期增殖阶段的关键物种。
米诺环素对活性污泥初期增殖阶段的影响
Impact of minocycline on the initial formation of activated sludge
-
摘要: 采用污泥连续培养的方式,分别考察了米诺环素在质量浓度为0、0.001、0.01、0.1、1和10 mg·L−1水平下对活性污泥初期增殖阶段的影响。结果表明,各组污泥增殖均能在20 d内进入平台期,且组间的稳态污泥量差异不显著 (P>0.05),表明活性污泥宏观生物量的增长与米诺环素浓度无关,但10 mg·L−1米诺环素的暴露可抑制污泥絮体的形成。细菌群落多样性的分析结果表明,1 mg·L−1的米诺环素即可导致活性污泥初期增殖阶段细菌多样性的降低,10 mg·L−1的米诺环素会选择性富集Flavobacterium菌属,使其丰度在第20天的活性污泥细菌群落中达到83.14%,可进一步降低形成的活性污泥细菌多样性。网络分析结果表明,Proteobacteria细菌是活性污泥初期增殖阶段的关键物种,低质量浓度米诺环素(0.001 mg·L−1和0.01 mg·L−1)可能促进了活性污泥细菌间的互作,而高质量浓度(1 mg·L−1和10 mg·L−1)暴露会显著降低互作网络的复杂度和稳定性。本研究结果可为含抗生素废水的高效生化处理提供参考。Abstract: Activated sludge (AS) was continuously incubated at the mass minocycline concentrations of 0, 0.001, 0.01, 0.1, 1 and 10 mg·L−1, respectively, and the effects of these concentrations on the initial formation of AS were investigated. The results showed that the biomass proliferation in 0~10 mg·L−1 minocycline treatments all entered the plateau stage within 20 days, and there were no significant differences (P> 0.05) in the steady-state sludge concentrations among the treatments, indicating that the growth of AS biomass was not delayed by the addition of minocycline, but the exposure of 10 mg·L−1 minocycline could lead to the failure of the formation of AS flocs. Miseq sequencing of 16S rRNA gene amplicons showed that the bacterial diversity of AS at its initial proliferation stage decreased in 1 mg·L−1 minocycline treatment, and 10 mg·L−1 minocycline exposure selectively enriched the genus Flavobacterium, and its abundance could reach 83.14% in the AS bacterial community at the end of the experiment, then the diversity of the formed AS bacterial community was further reduced. Network analysis showed that members of Proteobacteria were the key players in the initial formation of AS. Low mass concentration minocycline (0.001 and 0.01 mg·L−1) might promote the interaction between different modules of AS bacterial communities, while high mass concentration (1 and 10 mg·L−1) exposure would significantly reduce the complexity and stability of AS network. The results of the study can provide a reference for the efficient biochemical treatment of antibiotic-containing wastewater.
-
Key words:
- minocycline /
- activated sludge /
- proliferation /
- community construction /
- network analysis
-
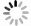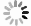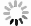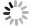
-
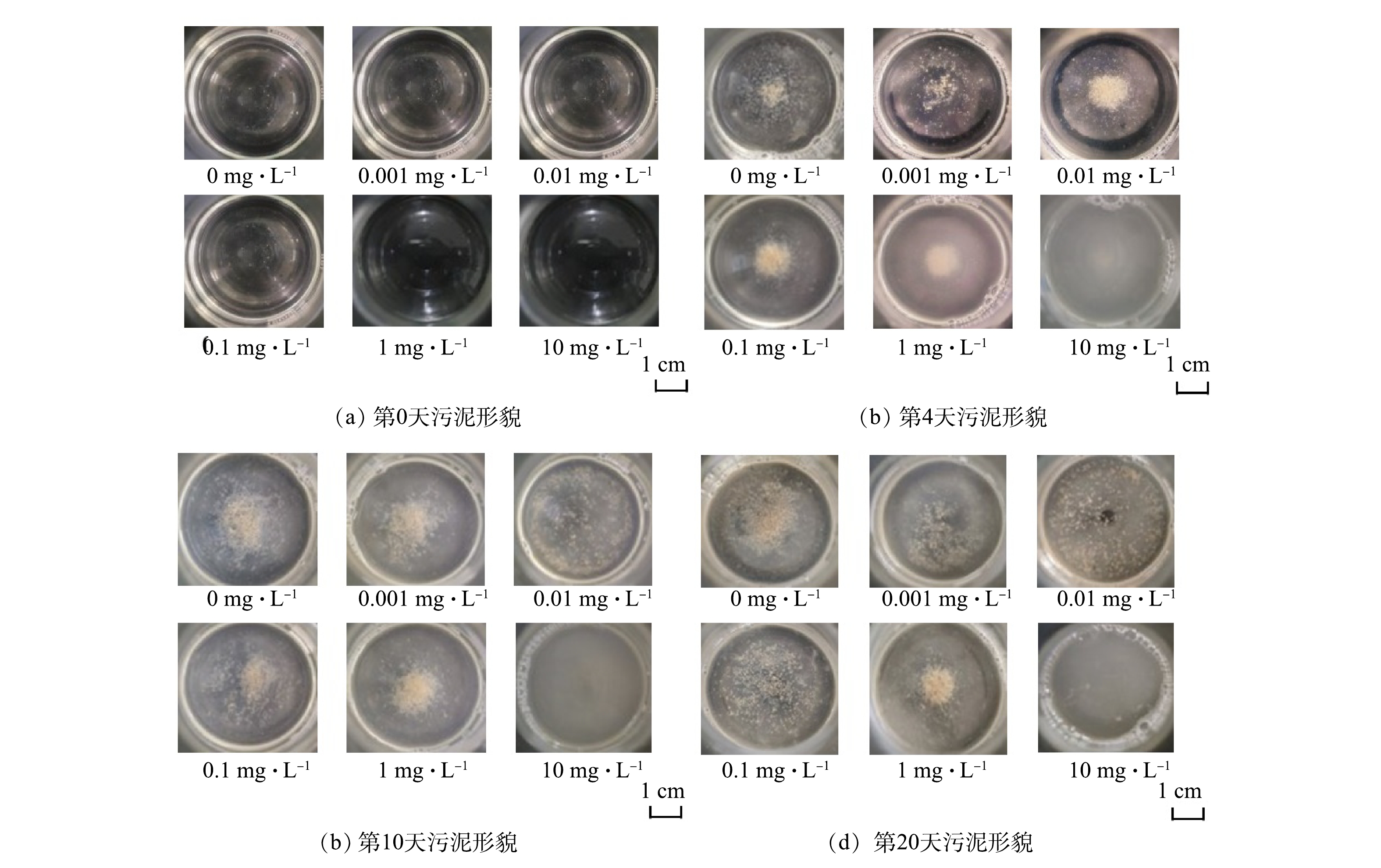
图 1 活性污泥初期增殖阶段宏观污泥形貌的变化
Figure 1. Changes of macroscopical sludge morphology during the initial formation of activated sludge under different minocycline stresses
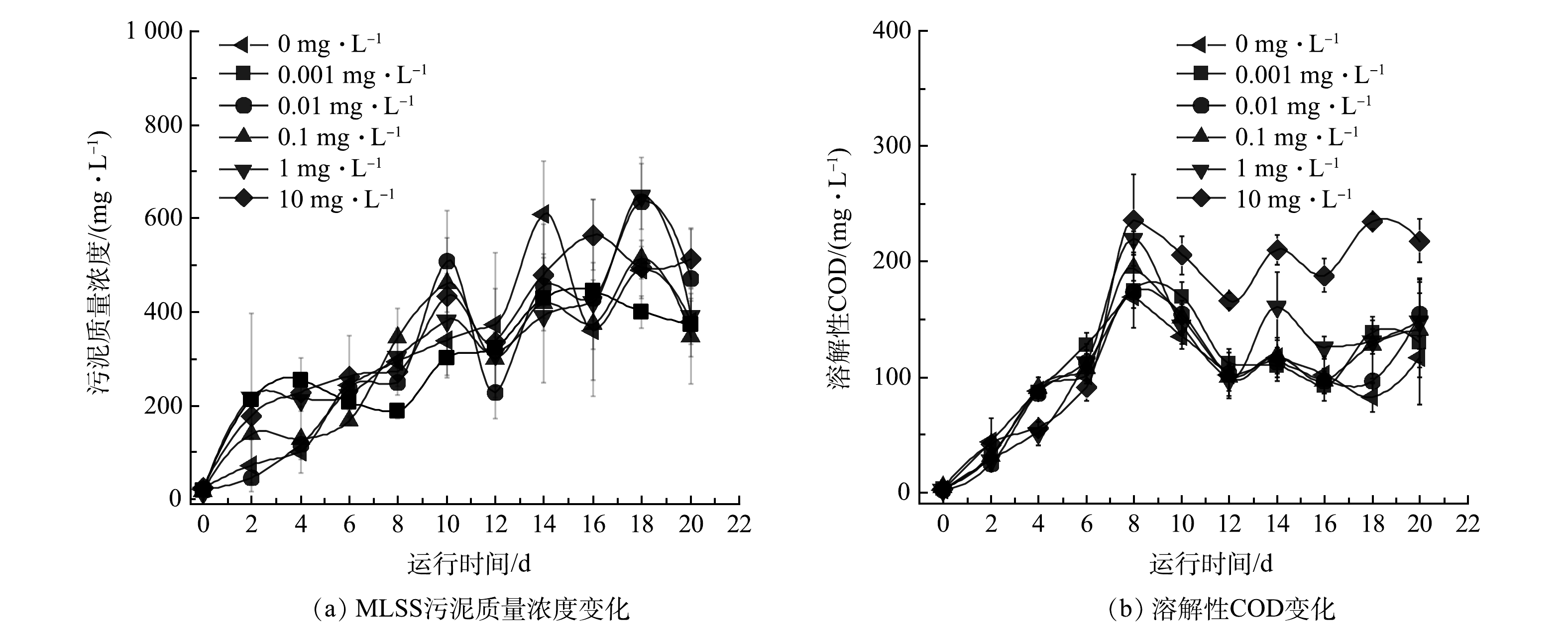
图 2 活性污泥初期增殖阶段各组生物量和代谢活性变化
Figure 2. Changes in biomass and metabolic activity of various groups during the initial proliferation phase of activated sludge
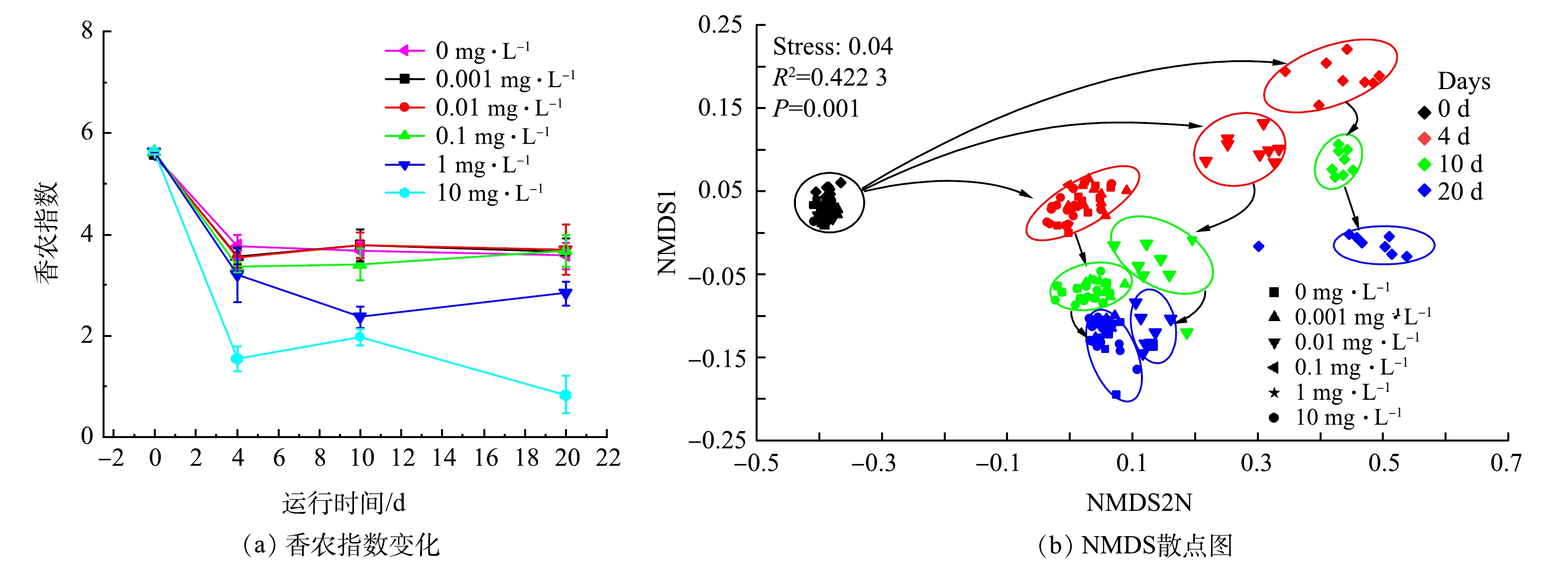
图 3 污泥初期增殖阶段细菌群落α和β多样性的变化
Figure 3. Changes of α-diversity and β-diversity of bacterial communities at the initial proliferation phase of sludge
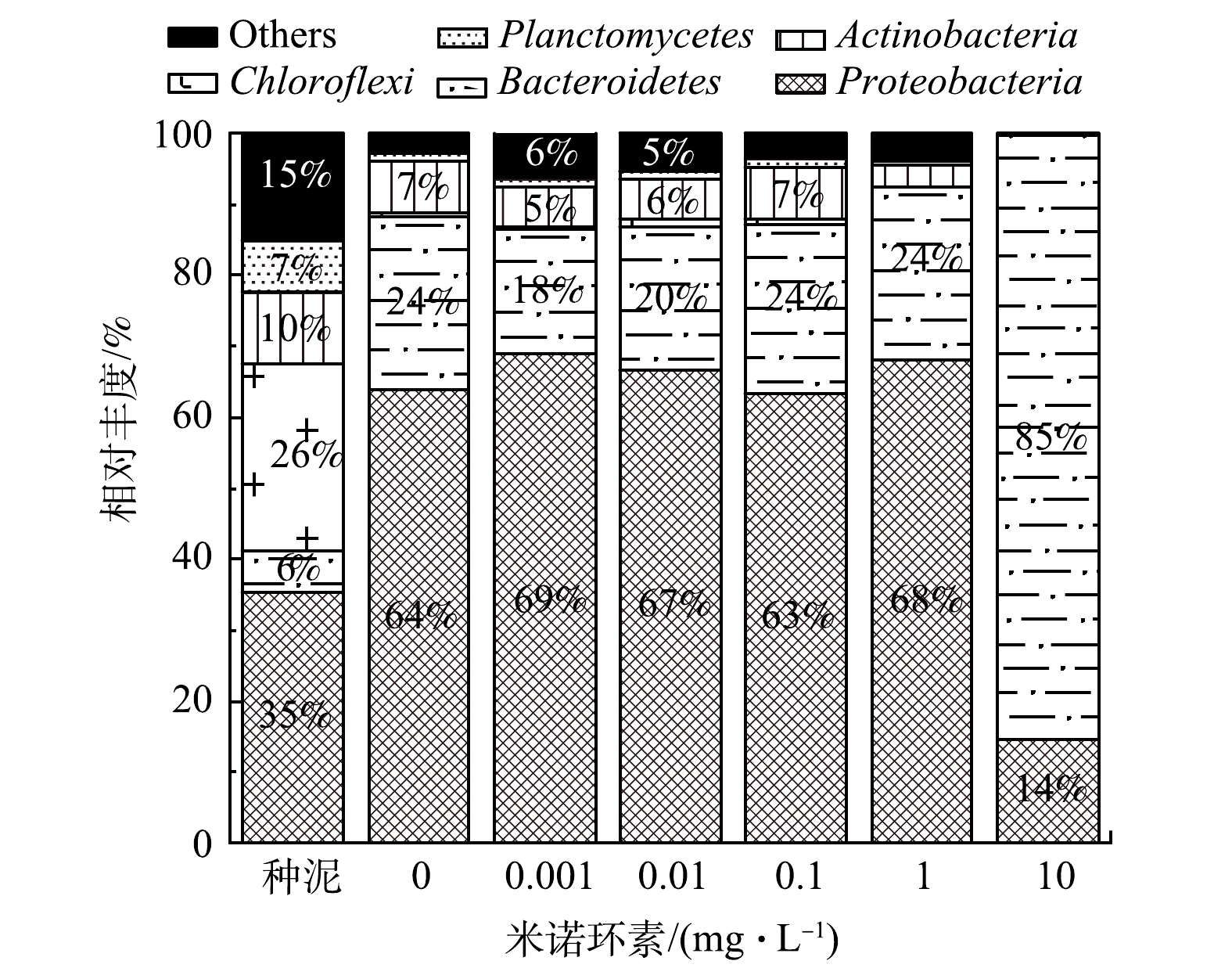
图 4 种泥及第20天各组细菌群落门水平的组成
Figure 4. Composition of the bacterial community at the phylum level for each group in seeding sludge and activated sludge on the 20th day
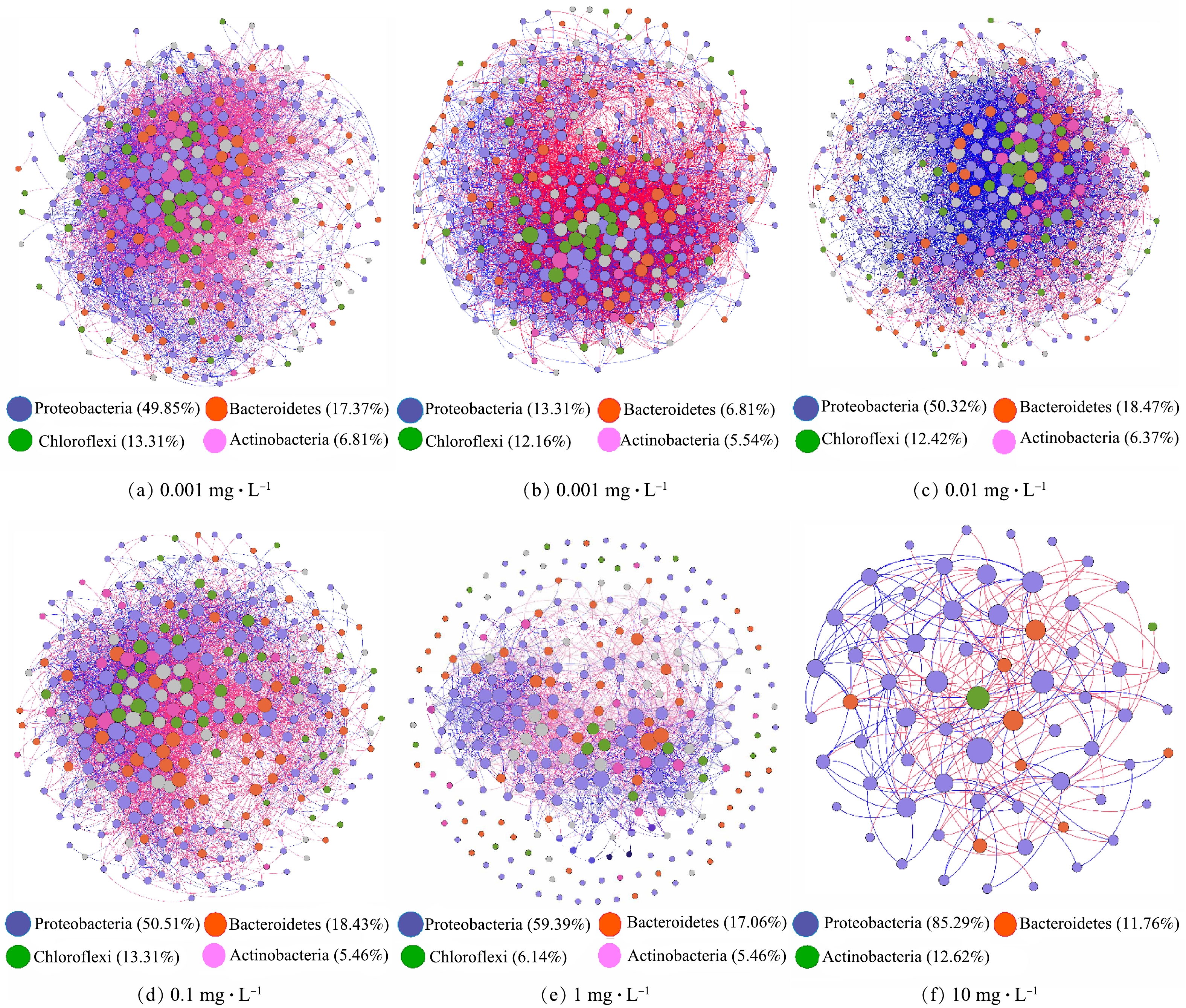
图 6 活性污泥初期增殖过程细菌群落的互作网络
Figure 6. Networks of bacterial communities during the initial formation of activated sludge
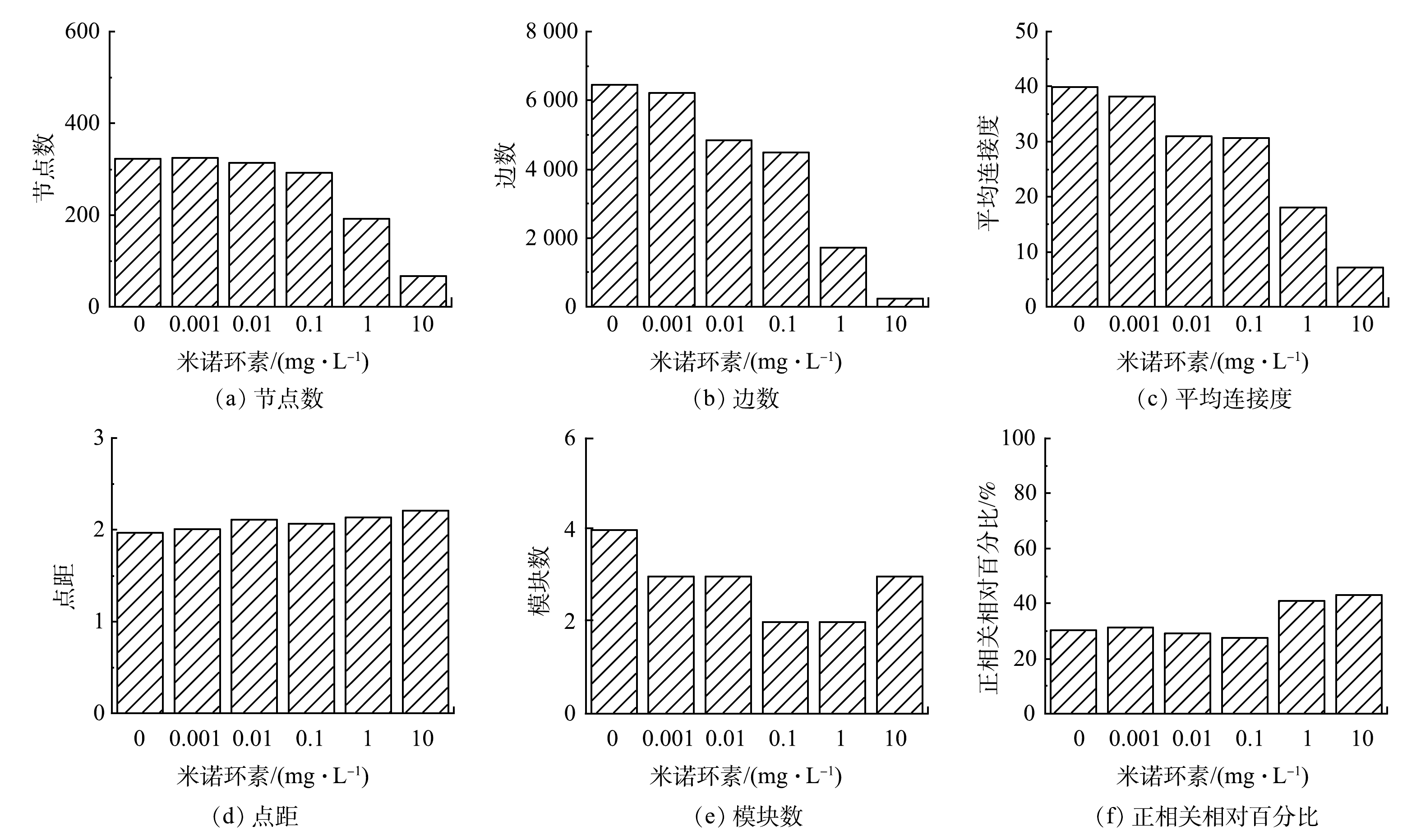
图 7 细菌互作网络的主要拓扑参数
Figure 7. Topological properties of bacterial co-occurrence networks
表 1 网络关键物种解析
Table 1. Key players during the initial formation of activated sludge revealed by network analysis
抗生素暴露
浓度/(mg·L−1)网络分析揭示的关键物种 网络中心点 模块中心点 连接节点 OTU Genus OTU Genus OTU/个 0 — — 2 889 norank_f__norank_o__PLTA13 10 432 Shinella 1 963 Paenarthrobacter 1 069 Propionicicella 0.001 — — 2 926 Hydrogenophaga 71 2 889 norank_f__norank _o__PLTA13 3 203 norank_f__A4b 0.01 3 142 Hyphomicrobium 4 820 Comamonas 51 794 unclassified_f_Burkholderiaceae 1 639 Pseudoxanth omonas 4 437 Comamonas 0.1 — – 1 639 Pseudoxanthomonas — 1 208 Leucobacter 775 unclassified_f__ Burkholderiaceae 613 unclassified_f__ Burkholderiaceae 1 — — 604 Brevundimonas — 1 — — 432 Shinella — 2 944 Acinetobacter 4 467 Sphingobacterium 3 142 Hyphomicrobium 4 863 Fluviicola 10 — — 4 349 Rheinheimera 2 注:*连接节点仅展示了OTU的总数量;其它节点仅展示OTU编号及所属分类;“–”代表无相关OTU。  下载: 导出CSV
下载: 导出CSV
-
[1] XIA S, DUAN L, SONG Y, et al. Bacterial community structure in geographically distributed biological wastewater treatment reactors[J]. Environmental Science & Technology, 2010, 44(19): 7391-7396. [2] WU L, NING D, ZHANG B, et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants[J]. Nature Microbiology, 2019, 4(7): 1183-1195. doi: 10.1038/s41564-019-0426-5 [3] ARDERN E, LOCKETTL W T. Experiments on the oxidation of sewage without the aid of filters[J]. Journal of the Society of Chemical Industry, 1914, 33(10): 523-539. doi: 10.1002/jctb.5000331005 [4] SHCHEGOLKOVA N M, KRASNOV G S, BELOVA A A, et al. Microbial community structure of activated sudge in treatment plants with different wastewater compositions[J]. Frontiers in Microbiology, 2016, 7: 90. [5] KIM M S, AHN S H, JEONG I J, et al. Water quality drives the regional patterns of an algal metacommunity in interconnected lakes[J]. Scientific Reports, 2021, 11(1): 1-9. doi: 10.1038/s41598-020-79139-8 [6] BOSSIER P, VERSTRAETE W. Triggers for microbial aggregation in activated sludge?[J]. Applied Microbiology Biotechnology, 1996, 45(1): 1-6. [7] 李培睿, 杨天佑, 李宗义, 等. 活性污泥凝絮体的形成过程研究[J]. 河南师范大学学报:自然科学版, 2007, 35(1): 150-152. [8] GAO C-H, CAO H, CAI P, et al. The initial inoculation ratio regulates bacterial coculture interactions and metabolic capacity[J]. The ISME Journal, 2020, 15(1): 29-40. [9] RATZKE C, BARRERE J, GORE J J, et al. Strength of species interactions determines biodiversity and stability in microbial communities[J]. Nature Ecology, 2020, 4(3): 376-383. doi: 10.1038/s41559-020-1099-4 [10] BURKE V, RICHTER D, GRESKOWIAK J, et al. Occurrence of antibiotics in surface and groundwater of a drinking water catchment area in Germany[J]. Water Environment Research, 2016, 88(7): 652-659. doi: 10.2175/106143016X14609975746604 [11] BEN W, ZHU B, YUAN X, et al. Occurrence, removal and risk of organic micropollutants in wastewater treatment plants across China: Comparison of wastewater treatment processes[J]. Water Research, 2018, 130: 38-46. doi: 10.1016/j.watres.2017.11.057 [12] BEHERA S K, KIM H W, OH J-E, et al. Occurrence and removal of antibiotics, hormones and several other pharmaceuticals in wastewater treatment plants of the largest industrial city of Korea[J]. Science of the Total Environment, 2011, 409(20): 4351-4360. doi: 10.1016/j.scitotenv.2011.07.015 [13] ARIKAN O A, RICE C, CODLING E. Occurrence of antibiotics and hormones in a major agricultural watershed[J]. Desalination, 2008, 226(1/2/3): 121-133. [14] HE L X, HE L Y, GAO F Z, et al. Antibiotics, antibiotic resistance genes and microbial community in grouper mariculture[J]. Science of the Total Environment, 2021, 808: 152042. [15] MA W L, QI R, ZHANG Y, et al. Performance of a successive hydrolysis, denitrification and nitrification system for simultaneous removal of COD and nitrogen from terramycin production wastewater[J]. Biochemical Engineering Journal, 2009, 45(1): 30-34. doi: 10.1016/j.bej.2009.02.001 [16] EKWANZALA M D, LEHUTSO R F, KASONGA T K, et al. Environmental dissemination of selected antibiotics from hospital wastewater to the aquatic environment[J]. Antibiotics, 2020, 9(7): 431. doi: 10.3390/antibiotics9070431 [17] YAO S, YE J, YANG Q, et al. Occurrence and removal of antibiotics, antibiotic resistance genes, and bacterial communities in hospital wastewater[J]. Environmental Science and Pollution Research International, 2021, 28(40): 57321-57333. doi: 10.1007/s11356-021-14735-3 [18] LINARES J F, GUSTAFSSON I, BAQUERO F, et al. Antibiotics as intermicrobial signaling agents instead of weapons[J]. Proceedings of the National Academy of Sciences, 2006, 103(51): 19484-19489. doi: 10.1073/pnas.0608949103 [19] LIU H, YANG Y K, SUN H F, et al. Effect of tetracycline on microbial community structure associated with enhanced biological N&P removal in sequencing batch reactor[J]. Bioresource Technology, 2018, 256: 414-420. doi: 10.1016/j.biortech.2018.02.051 [20] MA W, YANG M, WANG J, et al. Treatment of antibiotics wastewater utilizing successive hydrolysis, denitrification and nitrification[J]. Environmental Technology, 2002, 23(6): 685-694. doi: 10.1080/09593330.2002.9619253 [21] QIAO M, YING G-G, SINGER A C, et al. Review of antibiotic resistance in China and its environment[J]. Environment International, 2018, 110: 160-172. doi: 10.1016/j.envint.2017.10.016 [22] 国家环境保护总局. 水和废水监测分析方法-第4版[M]. 中国环境科学出版社, 2002. [23] 张滢楹, 耿金菊, 任洪强, 等. 环境浓度抗生素选择性压力改变活性污泥微生物群落结构[J]. 生态毒理学报, 2015(5): 66-74. [24] 陈燕, 刘国华, 范强, 等. 活性污泥法中细菌多样性综述[J]. 环境保护科学, 2015, 41(04): 70-78. doi: 10.3969/j.issn.1004-6216.2015.04.015 [25] 周梦娟, 缪恒锋, 陆震明, 等. 碳源对反硝化细菌的反硝化速率和群落结构的影响[J]. 环境科学研究, 2018, 31(12): 2047-2054. [26] TIAN Z, ZHANG Y, YANG M. Chronic impacts of oxytetracycline on mesophilic anaerobic digestion of excess sludge: inhibition of hydrolytic acidification and enrichment of antibiotic resistome[J]. Environmental Pollution, 2018, 238: 1017-1026. doi: 10.1016/j.envpol.2018.02.023 [27] TIAN Z, PALOMO A, ZHANG H, et al. Minimum influent concentrations of oxytetracycline, streptomycin and spiramycin in selecting antibiotic resistance in biofilm type wastewater treatment systems[J]. Science of the Total Environment, 2020, 720: 137531. doi: 10.1016/j.scitotenv.2020.137531 [28] ZHANG T, SHAO M-F, YE L. 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants[J]. The ISME Journal, 2012, 6(6): 1137-1147. doi: 10.1038/ismej.2011.188 [29] WAGNER M, LOY A, NOGUEIRAL R, et al. Microbial community composition and function in wastewater treatment plants[J]. Antonie Van Leeuwenhoek, 2002, 81(1): 665-680. [30] YANG C, ZHANG W, LIU R, et al. Phylogenetic diversity and metabolic potential of activated sludge microbial communities in full-scale wastewater treatment plants[J]. Environmental Science & Technology, 2011, 45(17): 7408-7415. [31] YU K, ZHANG T. Metagenomic and metatranscriptomic analysis of microbial community structure and gene expression of activated sludge[J]. PLoS One, 2012, 7(5): e38183. doi: 10.1371/journal.pone.0038183 [32] MALLON C A, VAN ELSAS J D, SALLES J F. Microbial invasions: the process, patterns, and mechanisms[J]. Trends in Microbiology, 2015, 23(11): 719-729. doi: 10.1016/j.tim.2015.07.013 [33] BERNARDET J F, BOWMAN J P. The genus Flavobacterium[J]. The Prokaryotes, 2006, 7: 481-531. [34] KIRCHMAN D L. The ecology of Cytophaga-Flavobacteria in aquatic environments[J]. FEMS Microbiol Ecology, 2002, 39(2): 91-100. [35] TIAN Z, LIU R, ZHANG H, et al. Developmental dynamics of antibiotic resistome in aerobic biofilm microbiota treating wastewater under stepwise increasing tigecycline concentrations[J]. Environment International, 2019, 131: 105008. doi: 10.1016/j.envint.2019.105008 [36] THAKER M, SPANOGIANNOPOULOS P, WRIGHT G D. The tetracycline resistome[J]. Cellular and Molecular Life Sciences, 2010, 67(3): 419-431. doi: 10.1007/s00018-009-0172-6 [37] DENG Y, JIANG Y H, YANG Y, et al. Molecular ecological network analyses[J]. BMC Bioinformatics, 2012, 13: 113. doi: 10.1186/1471-2105-13-113 [38] WANG S, WANG X, HAN X, et al. Higher precipitation strengthens the microbial interactions in semi‐arid grassland soils[J]. Global Ecology and Biogeography, 2018, 27(5): 570-580. doi: 10.1111/geb.12718 [39] HERNANDEZ D J, DAVID A S, MENGES E S, et al. Environmental stress destabilizes microbial networks[J]. The ISME Journal, 2021, 15(6): 1722-1734. doi: 10.1038/s41396-020-00882-x [40] 杜雄峰, 厉舒祯, 冯凯, 等. 农牧交错带草地土壤剖面微生物总量、多样性和互作网络的垂直分布特征[J]. 微生物学通报, 2020, 47(9): 2789-2806. [41] LAYTON A, KARANTH P, LAJOIE C, et al. Quantification of Hyphomicrobium populations in activated sludge from an industrial wastewater treatment system as determined by 16S rRNA analysis[J]. Applied and Environmental Microbiology, 2000, 66(3): 1167-1174. doi: 10.1128/AEM.66.3.1167-1174.2000 [42] KLOOS K, FESEFELDT A, GLIESCHE C G, et al. DNA-probing indicates the occurrence of denitrification and nitrogen fixation genes in Hyphomicrobium. Distribution of denitrifying and nitrogen fixing isolates of Hyphomicrobium in a sewage treatment plant[J]. FEMS Microbiology Ecology, 1995, 18(3): 205-213. doi: 10.1111/j.1574-6941.1995.tb00177.x [43] OLESEN J M, BASCOMPTE J, DUPONT Y L, et al. The modularity of pollination networks[J]. Proceedings of the National Academy of Sciences, 2007, 104(50): 19891-19896. doi: 10.1073/pnas.0706375104 -
 点击查看大图
点击查看大图
计量
- 文章访问数: 4251
- HTML全文浏览数: 4251
- PDF下载数: 84
- 施引文献: 0
