


-
硝化反应是自然界氮循环的关键环节,在氮生物地球化学循环中发挥着重要作用。硝化作用是氨氮(NH4+)首先被氨氧化细菌(AOB)或古菌(AOA)氧化为亚硝态氮(NO2−),进而被亚硝酸盐氧化菌(NOB)氧化为硝态氮(NO3−)的生化反应过程。在过去100多年中,众多学者研究了氨氧化反应过程及AOB菌群的生理特性,而亚硝酸盐氧化及NOB菌群的生理特性并未引起广泛重视。这主要是由于,在传统生物硝化过程中,氨氧化反应是硝化过程的限速步骤,相对于氨氧化反应,亚硝酸盐氧化反应较容易发生[1]。但短程生物脱氮技术的发现,通过不匹配AOB和NOB两类菌属的生理特性差异,选择性抑制NOB菌属的活性,可作为短程生物脱氮的有效途径,因此,众多学者开始关注NOB的菌群特性研究。
NOB属于化能自养型细菌,为专性好氧菌,可利用无机碳化合物,如CO2、CO32−、HCO3−等作为碳源,利用NO2−作为氮源,在NO2−氧化过程中以O2作为最终电子受体,以获得生长所需要的能量。NOB氧化NO2−产生的能量仅有2%~11%用于细胞增殖,因此,NOB能量利用率不高,生长较缓慢,导致其在自然生态系统中的比例较低(4%~5%)[2]。目前,已鉴定出来的NOB菌属主要包括硝化杆菌属(Nitrobacter)、硝化螺菌属(Nitrospira)、硝化球菌属(Nitrococuus)、硝化刺菌属(Nitrospina)、Nitrotoga属、Candidatus Nitromaritima属和Nitrolancea属7类菌属[3]。其中,硝化杆菌属(Nitrobacter)和硝化螺菌属(Nitrospira)是NOB的最典型代表菌属,广泛分布于土壤、淡水、海水及城市污水处理厂活性污泥中。
近10年来,高通量测序技术被广泛用于自然生态系统中微生物群落特性的研究中,能够系统深入地解析、刻画微生物组,通过核酸序列的同源性和差异反映群落的多样性、组成谱和功能谱,并对各种微生物组进行大规模并行比较、关联分析,从而有助于深入探讨它们对于整个生态系统的重要作用,更全面地研究“微生物组-环境/宿主”之间的相互作用机制。
本研究针对富集NOB菌属活性污泥系统,通过SEM技术探究了NOB菌属的形态特征,进而采用宏基因组学技术研究了生化系统内活性污泥的微生物组成及优势物种关联网络关系,探究了NOB菌属的生理生化、代谢活性和行为功能等多方面特性,为自然生态系统氮循环、短程生物脱氮技术提供微生物学理论支持。
-
实验采用人工模拟废水,其进水成分为:10~700 mg·L−1 NaNO2(通过投加不同的NaNO2溶液控制初始的NO2−浓度)、0.4 gNaHCO3、1 gKH2PO4、1.31 gK2HPO4以及0.9 mL微量元素。每升微量元素中:1.25 gEDTA、0.55 gZnSO4·7 H2O,、0.4 gCoCl2·6 H2O、1.275 gMnCl2·4 H2O、0.4 gCuSO4·4 H2O、0.05 gNa2MoO4·2 H2O、1.375 gCaCl2·2 H2O、1.25 gFeCl3·6 H2O、44.4 gMgSO4·7 H2O。接种污泥来自兰州西固兰炼污水处理厂氧化沟好氧段,该污泥具有良好脱氮除磷性能,混合液悬浮固体浓度为3 300 mg·L−1。
实验采用SBR反应器,其有效容积为7 L,控制反应器内混合液反应条件为,溶解氧4~4.5 mg·L−1、温度25±1 °C、pH 7.5±0.5。此外,采用WTW-Multi3420分析仪实时监测硝化反应终点。SBR典型运行周期为:瞬时进水(1 min)、曝气(时间随周期变化)、静置沉淀(60 min)和排水(5 min)。实验共运行140 d(420个周期),进水亚硝酸盐氮质量浓度从10 mg·L−1起步并以50 mg·L−1的梯度逐步提高至700 mg·L−1。
-
NO- 2-N、NO- 3-N、及悬浮固体浓度均采用国家标准方法测定[4-5];pH和溶解氧通过WTW-Multi3420进行实时监测。
-
污泥样品细菌基因组 DNA 采用DNeasyPowerSoil Kit试剂盒提取,并通过0.8%琼脂糖凝胶电泳进行DNA质量检测,同时采用紫外分光光度计对DNA的浓度和纯度进行量化。以细菌16S rRNA的V4-V5区为靶点,进行PCR扩增。扩增引物序列:F为515F(GTGCCAGCMGCCGCGGTAA),R为907R(CCGTCAATTCMTTTRAGTTT)。PCR扩增反应条件为:95 °C预加热2 min;随后进行25周期的扩增反应(95 °C变性30 s、55 °C退火30 s、72 °C延伸40 s);终止后在72 °C下保温10 min,反应液于−80 ℃保存。委托上海派森诺生物科技有限公司进行Illumina MiSeq高通量测序。
-
宏基因组物种注释和丰度分析:1)对高通量测序得到的原始序列进行质量筛查,获取高质量数据集(Clean data);2)对高质量序列进行序列拼接组装,构建宏基因组Contigs和 Scaffolds序列集,并将序列与 NCBI-NT 分析库中的真菌、细菌、古菌和病毒序列进行BLASTN比对(设定E<0.001);3)通过MEGAN[6]利用“最低共同祖先”算法[7]为目标序列的物种分类注释信息;4)结合Saffolds/Scaftigs序列在各组中的丰度数据,获得样本在系统分类等级上的相对丰度表,并进行基因预测,获得非冗余蛋白序列集;5)对蛋白序进行功能注释,获得功能类群丰度谱、代谢通路富集分析等。
-
絮状污泥电镜样品制备方法:1)取污泥混合液于1.5 mL的离心管中,以4 000 r·min−1离心5 min,将上清液去除,加入2.5%戊二醛固定,并置于4 °C冰箱中1.5 h;2)用0.1 mol·L−1的磷酸缓冲液冲洗3次,每次10 min;3)分别用质量分数50%,70%,80%,90%的乙醇进行脱水,每次10~15 min,再以100%的乙醇脱水3次,每次10~15 min;4)100%乙醇∶乙酸异戊酯= 1∶1、纯乙酸异戊酯各置换,每次15 min;5)用HCP22 (H ITACH I)型临界点干燥仪对样品进行干燥;6)在样品表面镀1 500 nm金属膜(金或铂膜)完成预处理。采用扫描电镜(JSM-5 600LV,日本)进行观察并进行能谱分析。
-
为了探究NOB富集系统内微生物主要形态特征,SBR生化系统稳定运行至第120天时,取活性污泥样品采用SEM对其表面形态进行表征(图1)。结果表明,微生物以杆状菌和椭球状菌为主[(0.5~0.8)×(1.0~2.0)],且活性污泥表面形状规则,界限清晰。此外,大量的菌体大都以单体形式存在,但又相互交织在一体,形成了网状的微生物系统。基于相关文献报道的NOB菌属微生物的形态特征,Nitrobacter呈短杆状,Nitrospira呈螺旋状,Nitrococuus呈球状,Nitrospina呈长杆状[8]。
-
基于活性污泥样品微生物在门分类水平群落分布(图2(a)),可以看出,微生物群落在门分类水平上多样性丰富,共检测出19个门。其中相对丰度较高(>1.0%)的门主要包括:变形菌门(Proteobacteria)(82.2%)、放线菌门(Actinobacteria)(4.5%)、拟杆菌门(Bacteroidetes)(4.5%)、厚壁菌门(Firmicutes)(2.1%)、芽单胞菌门(Gemmationadetes)(1.6%)和疣微菌门(Verrucomicrobia)(1.6%),这6类菌门的相对丰度总和约占总生物量的96.5%。其它相对丰度较低(<1.0%)的菌门包括:异常球菌-栖热菌门(Deinococcus-Thermus)(0.9%)、浮霉菌门(Planctomycetes)(0.8%)、绿弯菌门(Chloroflexi)(0.5%)等。NOB的代表性菌属中的Nitrobacter和Nitrospira分别属于Proteobacteria和硝化螺旋菌门(Nitrospirae)。Proteobacteria是自然生态系统细菌界中最大的门,也是活性污泥生化系统中最主要的功能菌群,其相对丰度约为27.5%~65.0%[9-10]。本研究中,Proteobacteria相对丰度(82.2%)高于已有的文献报道值。然而,杨浩等[11]在典型集雨人饮地区窖水微生物群落多样性及差异解析中发现,窖水水样中微生物在门水平上的群落结构中最丰富的是Bacteroidetes(39.1%),其次为Proteobacteria(29.2%)。宁高阳等[12]发现,碳氮比(C/N)能够显著影响Proteobacteria的相对丰度,在C/N为0时,Proteobacteria的相对丰度最低(40.7%),而C/N为5时,Proteobacteria的相对丰度最高(65.2%)。此外,Proteobacteria可被进一步划分为5个纲,分别为α、β、γ、δ和ε。其中,α-、β-、γ-和δ-Proteobacteria普遍共存于营养物去除生化系统中,且β-Proteobacteria是最主导的菌纲[13-14]。然而,本研究中α-、β-、γ-和δ-Proteobacteria的相对丰度分别为58.3%、16.1%、5.9%和1.9%。α-和β-Proteobacteria是较为优势的菌纲,这2类菌纲包括较多的好氧和兼性菌,可以代谢NO2−-N无机化合物。
图2(b)为微生物属水平上物种的组成及相对丰度。硝化杆菌属(Nitrobacter)、中慢生根瘤菌属(Mesorhizobium)、红长命菌属(Rubrivivax)、慢生根菌属(Bradyrhizobium)、伯克氏菌属(Burkholderia)、芽单胞菌属(Gemmatimonas)和丰祐菌属(Opitutus)等相对丰度>1.0%菌群有15属,共占总生物量的60.7%。此外,相对丰度小于<1.0%的菌属主要有链球菌属(Streptococcus)、噬酸菌属(Acidovorax)和鞘氨酸单胞菌属(Sphingomonas)等5属,共占总生物量的4.5%,共同构成了微生物在属水平上的群落结构。在这些微生物菌属中,Nitrobacter、Mesorhizobium和Rubrivivax是占总生物量比例较大的主导优势菌属,平均相对丰度分别为37.8%、3.9%和2.6%。其它微生物菌属的相对丰度总和占了全部生物量的34.9%,也是生化系统中微生物组成的重要部分。
本实验检测出3种NOB菌属,分别为硝化杆菌属(Nitrobacter)、硝化螺菌属(Nitrospira)和硝化刺菌属(Nitrospina)。其中,Nitrobacter是活性污泥系统中最优势菌属(相对丰度为37.8%),而Nitrospira和Nitrospina的相对丰度较低,分别仅为0.03%和0.05%。可见,以NaNO2为唯一氮源和能源,控制运行条件,逐步提高NO2−浓度梯度,可实现NOB种群的富集和优化,并使Nitrobacter成为NOB优势菌属。
有研究表明,Nitrobacter和Nitrospira是生物脱氮系统中最为代表性的NOB菌属[15],但2类菌属的生理生化特性有显著差异。在高NO2−基质底物浓度、高溶解氧条件下,Nitrobacter含量远高于Nitrospira,即有利于Nitrobacter的增殖[16-17]。本实验NOB富集过程中NO2−浓度从10 mg·L−1逐步提高至700 mg·L−1,且运行过程DO浓度维持在4.0 mg·L−1以上,有利于Nitrobacter的培养。相对于Nitrobacter,Nitrospira具有生长速率缓慢、高产率和高底物亲和力等生理生化特性,适宜在基质浓度限制的寡营养环境中生长[18]。姚倩等[19]采用SBR反应器,在NO2−浓度始终低于2 mg·L−1、温度为26 ℃、DO为3~4 mg·L−1的条件下,成功富集了以Nitrospira(相对丰度75%)为主的活性污泥。基于上述分析,本实验的运行效果与SEM及高通量微生物多样性测序结果相一致,充分表明富集培养后SBR系统污泥微生物中NOB菌群中Nitrobacter占绝对优势地位。
-
微生物Spearman关联网络分析是基于OTU在活性污泥样品中的相对丰度分布,利用相关性分析探寻彼此之间呈现正相关或负相关的微生物类群,进而做出关联网络图[20]。通过网络关联分析,找寻群落成员共同出现(Co-occurrence)或彼此排斥(Co-exclusion)的相互作用模式,从而推断不同微生物类群之间可能的相互“协作”或“竞争”关系[21]。使用R语言,计算活性污泥系统丰度位于前50位优势属之间的Spearman等级相关系数,对其中|Spearman’ρ|>0.8且p<0.01的相关优势属构建关联网络,导入Cytoscape进行数据可视化(图3)。图中颜色相同的节点代表这类微生物属于同一个属分类水平,且节点越大,表明该微生物在微生物菌群中的相对丰度越高。节点连线越多,说明系统微生物菌群与该目标微生物之间联系越密切。节点之间的连线表明两者之间存在相关性(红线代表正相关,绿线代表负相关)。
在优势物种关联网络图中,左圆和右圆内微生物菌群均为呈正相关性的菌种,两圆之间均为呈现负相关性的菌种。其中,Nitrobacter中的维氏硝酸杆菌种(Nitrobacter winogradskyi)、汉氏硝酸杆菌种(Nitrobacter hamburgensis)、红假单胞菌种(Rhodopseudomonas)、中慢生根瘤菌种(Mesorhizobium)、生丝微菌种(Hyphomicrobium)及短根瘤菌种(Brad yrhizobium)等表现为正相关关系;但与芽单胞菌种(Gemmatimonas)、纤丝菌种(Leptothrix)、链球菌种(Streptococcus)、分枝杆菌种(Ramlibacter)及寡养单胞菌种(Stenotrophomonas)呈现负相关关系。在所有菌种中,维氏硝酸杆菌种(Nitrobacter winogradskyi)的相对丰度最高,为33.0%,其次为汉氏硝酸杆菌种(Nitrobacter hamburgensis),相对丰度为4.5%,这两个菌种作为Nitrobacter进化树的重要分支,在活性污泥生物脱氮过程扮演重要的角色,它们之间具有较高的相似度(p<0.01)[22]。与Nitrobacter winogradskyi和Nitrobacter hamburgensis呈正相关的主要菌种包括,Mesorhizobium、Bradyrhizobium、Sinorhizobium、Rhodobacter和 Chelativorans等16个菌种。其中,Mesorhizobium与两类菌种的正相关性最强,Spearman’ρ达到0.999和0.998,这表明这些微生物的成员在系统中以协同作用(Co-occurrence)出现,完成NO2−的氧化。与Nitrobacter winogradskyi和Nitrobacter hamburgensis菌种呈负相关的菌种有Variovorax、Ralstonia、Truepera、Streptococcus和Stenotrophomonas等14个菌种。其中,Streptococcus与Variovorax和Ralstonia的负相关性最强,Spearman’ρ达到-0.999和-0.999,表明两类菌种与这些微生物的成员在系统中以彼此排斥(Co-exclusion)的相互作用模式共存,不利于NO2−的氧化。
-
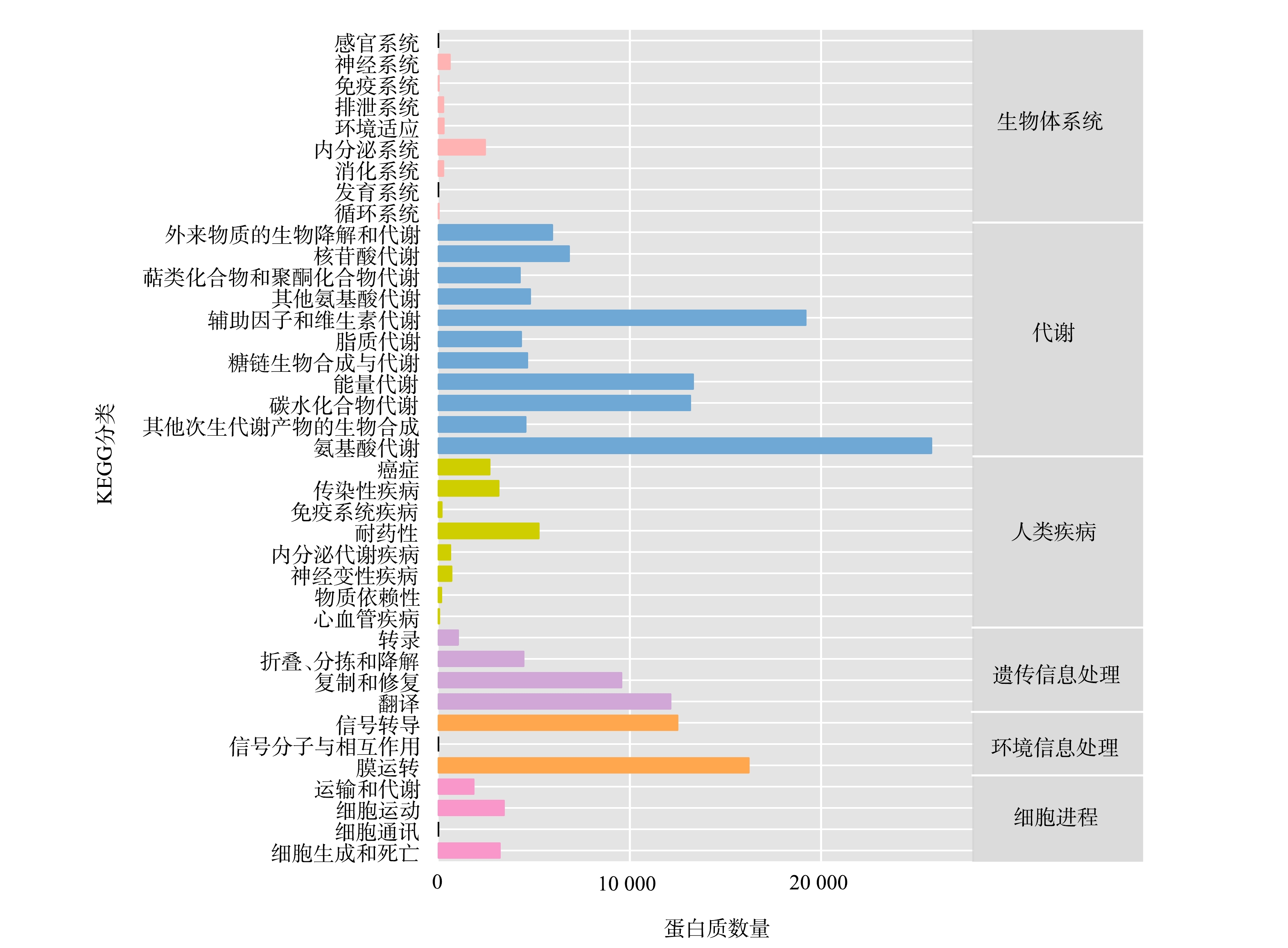
微生物宏基因组基因功能注释是将非冗余蛋白序列集与常用蛋白数据库比对,对各污泥样品中的基因功能进行注释分析。目前,应用最广泛的提供功能注释的数据库是KEGG代谢通路数据库。KEGG为生物代谢通路分析数据库,包括代谢、遗传信息处理、环境信息处理、细胞进程、生物体系统和人类疾病等6大类代谢通路。将蛋白序列和KEGG代谢通路数据库比对,对宏基因组预测得到的基因进行注释和分类。将上述非冗余蛋白序列集上传至KAAS[23]进行功能注释,对注释结果进行汇总,获取各等级的注释结果及对应的功能蛋白绝对丰度信息。
注释结果如图4所示。图中横坐标为蛋白序列注释到相应代谢通路的蛋白质数量,纵坐标左侧为KEGG第二等级代谢通路子功能,其所属的第一等级分类在右侧列出。在第一等级代谢通路中代谢、遗传信息处理、环境信息处理、细胞进程、生物体系统和人类疾病注释到的蛋白质数量分别为106 698、27 149、28 711、8 460、3 804和12 638个。其中,注释到代谢通路的蛋白质数量最多,占总蛋白数量的56.9%,其次为是环境信息处理(15.4%)和遗传信息处理(14.5%),其余3种通路的相应功能的蛋白丰度较低(2.0~6.7%)。新陈代谢功能模块中编码相应功能的基因在功能上的多样性和数量上均具有显著的优势性,反映了微生物新陈代谢是各类菌属与环境之间主导的物质和能量交换以及生物体内物质和能量的生命活动过程。
在第二等级代谢通路子功能水平上,代谢通路中注释到氨基酸代谢的蛋白数值最多,为25 756个(13.7%),其次为辅助因子和维生素代谢,其功能蛋白数量为19 196个(10.2%)。环境信息处理中注释到膜运转的蛋白数量为16 212个(8.6%)。注释到碳水化合物代谢、能量代谢、翻译和信号传导的蛋白质数量几乎处在一个相同的水平,均值为12 771个(6.8%)。基于KEGG的蛋白功能注释功能表明,活性污泥系统内具有相对特定功能的微生物种群。Nitrobacter菌属作为系统中最优势菌种,在将NO2−氧化成NO3−的生化反应过程中,进行着诸多活跃的与新陈代谢相关活动。
此外,在KEGG第四等级对应的代谢通路上获得各个KO直系同源基因簇的具体注释信息,将所有KO数据映射到KEGG的氮代谢通路图,获得系统与氮代谢有关的酶。在硝化、反硝化、同化硝酸盐还原和异化硝酸盐还原氮转化过程中,共获得18种氮代谢相关的基因(表1、图5)。其中,亚硝酸盐氧化还原酶Nxr(nxrA和nxrB基因)的相对丰度最高,为13.5%。Nxr是NOB的功能标记物,是亚硝酸盐氧化的关键酶,主要有细胞质型和细胞周质型2种类型,都属于络合物铁-硫钼酶家族,由α亚基(nxrA)、β亚基(nxrB)和γ亚基(nxrC)组成[24]。目前,nxrA和nxrB分别作为硝化杆菌(Nitrobacter)和硝化螺菌(Nitrospira)的功能基因和系统发育标记物被广泛应用[25-26],本实验过程所有样品中,均检测出nxrA和nxrB,两者的相对丰度分别为8.6%和4.9%,且在所有的基因中,nxrA的相对丰度最大,这也从功能基因层面证实了Nitrobacter是生化系统内的最优势菌属,亚硝酸盐氧化反应是系统内起主导作用的生化反应。
-
1) 基于16S rRNA宏基因组对活性污泥物种注释和丰度结果显示,Proteobacteria为活性污泥系统中最优势菌门(约占82.2%),硝化杆菌属(Nitrobacter)为最优势菌属(约占37.8%),其余菌属占比均低于5.0%,表明经过富集培养后SBR活性污泥系统微生物群落中NOB占绝对优势地位。
2) 微生物种水平优势物种关联网络表明维氏硝酸杆菌(Nitrobacter winogradskyi)和汉氏硝化细菌(Nitrobacter hamburgensis)相对丰度较高,分别为33.0%、4.5%,是Nitrobacter的主导物种,且两者呈现出显著正相关性。
3) 基于KEGG数据库蛋白功能注释分析结果表明,NOB富集系统进行着多种新陈代谢功能(56.9%)、环境信息处理(15.4%)以及遗传(14.5%)活动,且基于氮代谢过程相关酶的种类及丰度表明nxrA和nxrB是亚硝酸盐氧化的最优势功能基因,两者的相对丰度占全功能基因丰度总和的13.5%。
基于宏基因组学的亚硝酸盐氧化菌属的新陈代谢功能分析
Metabolic function analysis of nitrite oxidizing bacteria based on metagenomics
-
摘要: 为探究亚硝酸盐氧化菌(NOB)的菌群结构及其新陈代谢功能特性,在富集NOB菌属的基础上,基于宏基因组学技术探究了富集活性污泥系统内NOB微生物种群结构、相互作用关系、氮转化相关功能基因及蛋白质功能特性。结果表明,Nitrobacter为富集后活性污泥系统中最优势菌属,相对丰度为37.8%。微生物菌种之间表现出各种错综复杂的关联特征,其中,维氏硝酸杆菌种(Nitrobacter winogradskyi)和汉氏硝化菌种(Nitrobacter hamburgensis)呈现出显著正相关性。此外,新陈代谢是系统内各类菌属中主导的生命活动过程,亚硝酸盐氧化还原酶Nxr为生化系统内氮代谢的关键酶(占比13.5%)。本研究结果可为NOB菌属的富集培养提供参考。Abstract: In order to explore the community structure and metabolic function of nitrite oxidizing bacteria (NOB) in an activated sludge system, the microbial population structure, interaction relationship, nitrogen conversion related functional factors and protein functional characteristics in the NOB enriched activated sludge system were explored based on metagenomics technology. Results showed that Nitrobacter was the most abundant bacteria in the activated sludge system after enrichment, with relative abundance of 37.8%. Among them, Nitrobacter winogradskyi and Nitrobacter hamburgensis showed a strong positive correlation. In addition, metabolism was the most dominant process of all kinds of bacteria in the system, and Nitrite oxidoreductase Nxr was the key enzyme of nitrogen metabolism in biochemical system (accounting for 13.5%). The results of this study can provide a reference for the enrichment culture of NOB bacteria.
-
-

图 1 富集系统NOB活性污泥表面SEM分析
Figure 1. SEM analysis of enriched NOB activated sludge surface

图 3 种水平的优势微生物关联网络图
Figure 3. Association network of dominant microorganisms at species level
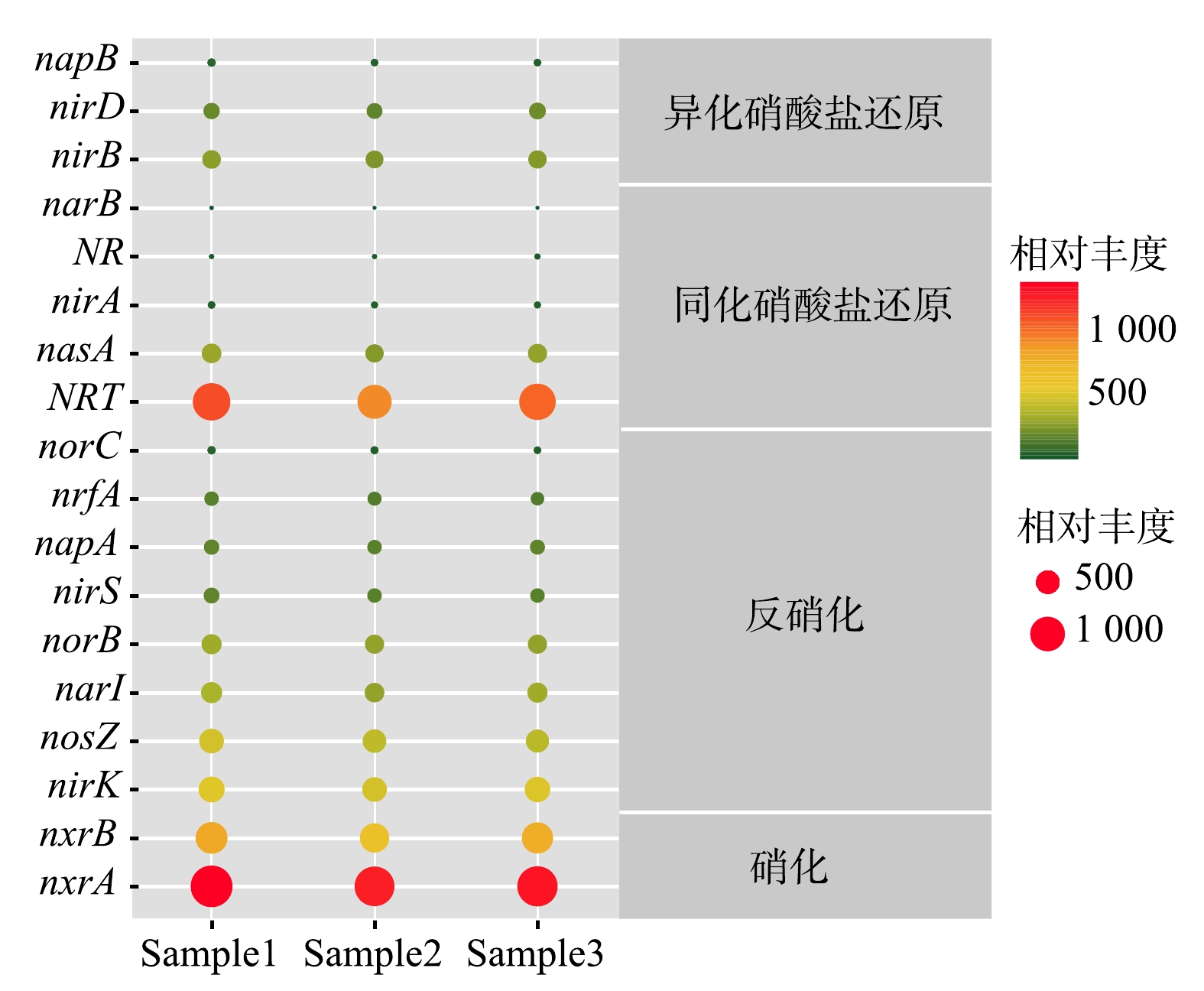
图 5 NOB富集系统氮循环功能基因相对丰度
Figure 5. NOB enrichment system nitrogen cycle gene annotation results
表 1 氮代谢过程相关酶的种类及丰度
Table 1. Types and abundances of enzymes related to nitrogen metabolism process
反应过程 相对丰度/% 相关酶 反硝化过程 14.3 nirK、nosZ、narI、norB、nirS、napA、nrfA、norC 硝化过程 13.5 nxrA、nxrB 异化硝酸盐还原过程 6.6 nirB、nirD、napB 同化硝酸盐还原过程 9.3 NRT、nasA、nirA、NR、narB  下载: 导出CSV
下载: 导出CSV
-
[1] TAYLOR A E, MYROLD D D, BOTTOMLEY P J. Temperature affects the kinetics of nitrite oxidation and nitrification coupling in four agricultural soils[J]. Soil Biology and Biochemistry, 2019, 136: 107523. doi: 10.1016/j.soilbio.2019.107523 [2] ZHANG L, SHEN Z, FANG W, et al. Composition of bacterial communities in municipal wastewater treatment plant[J]. Science of the Total Environment, 2019, 689: 1181-1191. doi: 10.1016/j.scitotenv.2019.06.432 [3] DAIMS H, S L, WAGNER M. A New Perspective on Microbes Formerly Known as Nitrite-Oxidizing Bacteria[J]. Trends in Microbiology, 2016: 699-712. [4] 国家环境保护局. 水和废水监测分析方法[M]. 4版. 北京: 中国环境科学出版社, 2002. [5] APHA. Standard methods for the examination of water and wastewater[J]. 20th ed. Washington, D. C. :American Public Health Association, 1998: 80-90. [6] HUSON D H, MITRA S, RUSCHEWEYH H J, et al. Integrative analysis of environmental sequences using MEGAN4[J]. Genome Research, 2011, 21(9): 1552-1560. doi: 10.1101/gr.120618.111 [7] HUSOM D H, AUCH A F, QI J, et al. MEGAN analysis of metagenomics data[J]. Genome Research, 2007, 17(3): 377-386. doi: 10.1101/gr.5969107 [8] 于雪. 温度、pH值和游离亚硝酸对亚硝酸盐氧化菌活性动力学及微生物种群结构影响研究[D]. 兰州: 兰州交通大学, 2019. [9] ZHANG T, SHAO M, YE L. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14sewage treatment plants[J]. The ISME Journal, 2011, 6(6): 1137-1147. [10] FANG D X, ZHAO G, XU X Y, et al. Microbial community structures and functions of wastewater treatment systems in plateau and cold regions[J]. Bioresource Technology, 2018, 249: 684. doi: 10.1016/j.biortech.2017.10.063 [11] 杨浩, 张国珍, 杨晓妮, 等. 典型集雨人饮地区窖水微生物群落多样性及差异解析[J]. 环境科学, 2017, 38(11): 4733-4746. [12] 2] 宁高阳, 牛永健, 李维维, 等. C/N对SBR生物脱氮硝化过程微生物结构的动态影响[J]. 中国环境科学, 2020, 40(05): 2053-2061. doi: 10.3969/j.issn.1000-6923.2020.05.023 [13] SUN H W, SHI W Y, CAI CH J, et al. Responses of microbial structures, functions, metabolic pathways and community interactions to different C/N ratios in aerobic nitrification[J]. Bioresource technology, 2020: 311. [14] PENG Y, ZHU G. Biological nitrogen removal with nitrification and denitrification via nitrite pathway[J]. Applied Microbiology and Biotechnology, 2006, 73(1): 15-26. doi: 10.1007/s00253-006-0534-z [15] CAO J S, ZHANG T, WU Y, et al. Correlations of nitrogen removal and core functional genera in full-scale wastewater treatment plants: Influences of different treatment processes and influent characteristics[J]. Bioresource Technology, 2020, 297: 122455. doi: 10.1016/j.biortech.2019.122455 [16] 董怡君, 王淑莹, 汪传新, 等. 亚硝酸盐氧化菌(NOB)的富集培养与其污泥特性分析[J]. 中国环境科学, 2013, 33(11): 1978-1983. [17] 包鹏, 王淑莹, 马斌, 等. 不同溶解氧间歇曝气对亚硝酸盐氧化菌的影响[J]. 中国环境科学, 2016, 36(9): 2696-2702. doi: 10.3969/j.issn.1000-6923.2016.09.024 [18] VIJAYAN A, JAVADRADHAN R, PILLAI D, et al. Nitrospira as versatile nitrifiers: Taxonomy, ecophysiology, genome characteristics, growth, and metabolic diversity[J]. Journal of Basic Microbiology, 2021(2). [19] 姚倩, 彭党聪, 赵俏迪, 等. 活性污泥中硝化螺菌(Nitrospira)的富集及其动力学参数[J]. 环境科学, 2017, 38(12): 5201-5207. [20] FAUST K, RAES J. Microbial interactions: from networks to models[J]. Nature Reviews Microbiology, 2012, 10(8): 538-550. doi: 10.1038/nrmicro2832 [21] JU F, ZHANG T. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant[J]. Isme Journal, 2015, 9(3): 683. doi: 10.1038/ismej.2014.162 [22] TESKE A, ALM E, REGAN J M, et al. Evolutionary relationships among ammonia- and nitrite-oxidizing bacteria[J]. Journal of Bacteriology, 1994, 176(21): 6623. doi: 10.1128/jb.176.21.6623-6630.1994 [23] MORIYA Y, ITOH M, OKUDA S, et al. KAAS: an automatic genome annotation and pathway reconstruction server[J]. Nucleic Acids Res, 2007, 35: W182-W185. doi: 10.1093/nar/gkm321 [24] PEATER M, MAIXNER F, BERRY D, et al. NxrB encoding the beta subunit of nitrite oxidoreductase as functional and phylogenetic marker for nitrite-oxidizing Nitrospira[J]. Environmental Microbiology, 2014, 16(10): 3055-3071. doi: 10.1111/1462-2920.12300 [25] LAI X S, ZHAO Y Q, PAN F X, et al. Enhanced optimal removal of nitrogen and organics from intermittently aerated vertical flow constructed wetlands: Relative COD/N ratios and microbial responses[J]. Chemosphere, 2020: 244. [26] YIN Y N, YANG CH, GU J, et al. Roles of nxrA-like oxidizers and nirS-like reducers in nitrite conversion during swine manure composting[J]. Bioresource Technology, 2019, 297: 122426. -
 点击查看大图
点击查看大图
图( 5) 表( 1)
计量
- 文章访问数: 5266
- HTML全文浏览数: 5266
- PDF下载数: 109
- 施引文献: 0
