



 下载:
下载:




-
纺织印染工业产生的难生物降解、毒性大、色度高和具有三致效应的印染废水量已呈现出逐年上升趋势[1-2]。若处理不当,排入天然水体的印染废水不仅会对水质、水生生物和生态系统造成严重影响,还可以通过食物链富集威胁人类健康。因此,对印染废水进行有效、合理地处理对保护水资源具有重要意义。
近年来,基于
${\rm{SO}}_4^ - \cdot $ 的高级氧化工艺因能够克服传统芬顿氧化体系具有的pH适用范围过窄[3]、活性物种氧化能力过弱[4]、存在时间过短[5]等难题而得到广泛运用。但其均相反应体系中仍存在金属离子难回收、容易产生污泥、二次污染严重等问题[6]。因此,开发新型的易回收、化学性质稳定、氧化能力强的固体催化剂并以其构建非均相反应体系成为现阶段该方法研究的热点和难点。在活化PMS的方法中,过渡金属活化法具有易操控、耗能小等特点。而过渡金属中,尤其是磁性铁基催化剂活化PMS降解染料一直是有机废水处理领域的研究热点。如何在铁基催化剂的基础上进一步提高Fe2+/Fe3+的转化效率也逐渐成为现阶段该技术手段的难点。本研究利用绿色可回收的铁基材料[7]、氧化还原能力优良的铈氧化物[8]以及具有共催化作用的二硫化钼[9],构建了二硫化钼负载磁性铈铁氧化物活化PMS降解橙黄II染料的体系,以期在获得高效的降解率的同时可以提高催化剂的回收利用性能,进而减少二次污染和资源浪费。
全文HTML

-
六水合硝酸铈(Ce(NO3)3·6H2O)、九水合硝酸铁((Fe(NO3)3·9H2O)、无水柠檬酸((C8H8O7)、氨水(NH3·H2O)、无水甲醇(CH4O)、硫脲(CH4N2S)、橙黄II(C16H11N2NaO4S)、四水合钼酸铵((NH4)6Mo7O24·4H2O)、过一硫酸氢钾(KHSO5·0.5KHSO4·0.5K2SO4)、叔丁醇(C4H10O)、苯酚(C6H6O)、高氯酸钠(NaClO4)、对苯醌(C6H4O2)、硫酸(H2SO4)、氢氧化钠(NaOH)均为分析纯,实验用水为超纯水。
-
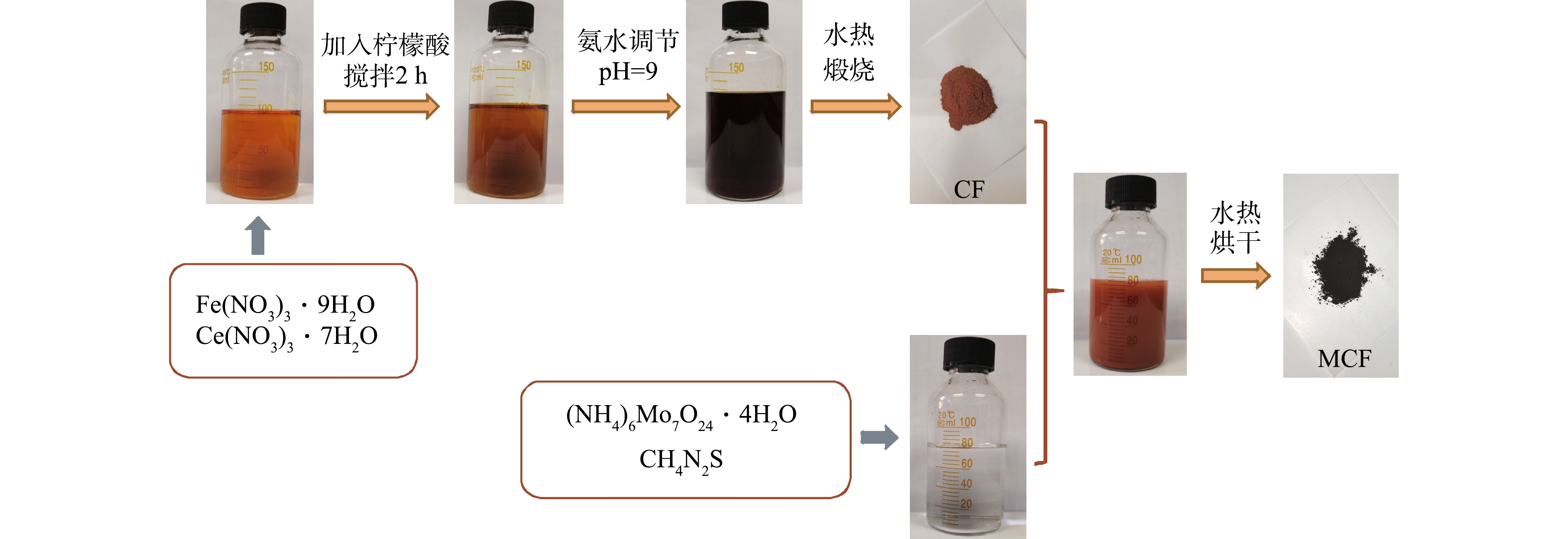
首先,称取一定量的Ce(NO3)3·6H2O和Fe(NO3)3·9H2O溶于90 mL超纯水中,再加入7.685 g C8H8O7,待其溶解后持续搅拌2 h并用NH3·H2O调节溶液pH至9;然后,将上述混合液转移至特氟龙反应釜中并在180℃下保持20 h,待反应釜冷却后,将固体产物用去离子水和无水乙醇分别洗涤数次,并在60℃下烘干,将烘干固体研磨成粉末,将粉末置于马弗炉中以一定温度煅烧6 h得到磁性铈铁氧化物(CF);最后,在水热法合成二硫化钼的过程[10]中加入CF制得二硫化钼-磁性铈铁氧化物复合材料,制备流程见图1。
-
采用扫描电子显微镜(SEM,JSM-7800,日本JEOL公司)和透射电子显微镜(TEM,JEM-2100F,日本JEOL公司)分析材料的形貌、晶型。采用X射线衍射仪(XRD,D8 ADVANCE,德国Bruker公司)定性分析材料的化学组成。采用Zeta电位仪(Zetasizer Nano ZS系列,英国Malvern公司)测定MCF在不同pH下的Zeta电位值。使用振动样品磁强计(VSM,EZ7,美国MicroSense公司)对材料的磁性强弱进行定量分析。反应溶液中溶出的铁离子和铈离子浓度采用电感耦合等离子原子发射光谱仪(ICP-OES,5800,美国Agilent公司)进行测定。采用电子顺磁共振波谱仪(EPR,A-300,德国Bruker公司)对反应过程中产生的活性物种进行识别。采用光电子能谱仪(XPS,ESCALAB 250Xi,美国Thermo公司)测定反应前后含氧物种含量的变化。
-
降解实验在恒温水浴振荡器中进行。将50 mL质量浓度为25 mg·L−1的AO7溶液和一定量的MCF置于血清瓶中振荡吸附30 min并取样,随后加入PMS启动氧化反应,并在氧化反应的不同时间段取样,所有样品取出后均立即加入等量甲醇淬灭。采用紫外-可见分光光度计在485 nm处测定样品吸光度。每次实验完成后利用抽滤装置回收催化剂,并通过数次乙醇和超纯水洗涤后烘干,所得催化剂再进行重复降解实验。
不同反应体系影响实验中催化剂投加量为0.6 g·L−1,PMS浓度为1 mmol·L−1。MCF投加量影响实验中PMS浓度为1 mmol·L−1。如无特殊说明,降解实验中MCF投加量为1.2 g·L−1,PMS为2 mmol·L−1,反应pH为初始值。采用甲醇和叔丁醇鉴识溶液中的·OH与
${\rm{SO}}_4^ - \cdot $ [11],选用苯酚分辨溶液中和材料表面的·OH与${\rm{SO}}_4^ - \cdot $ [12];通过高氯酸钠辨别反应中非自由基的反应机制[13];使用对苯醌和L-组氨酸鉴别反应中生成的$ \cdot {\rm{O}}_2^ - $ 和1O2[11, 14]。实验中采用准一级反应动力学模型(式(1))和二级动力学模型(式(2))对不同催化剂投加量的反应过程进行拟合。
式中:C为t时刻橙黄II的瞬时质量浓度,mg·L−1,C0为橙黄Ⅱ的初始质量浓度,mg·L−1;t为氧化降解时间,min;kobs为准一级反应速率常数,min−1;k2为二级反应速率常数,min−1。
1.1. 实验原料
1.2. 催化剂制备方法
1.3. 表征方法
1.4. 实验与分析方法



-
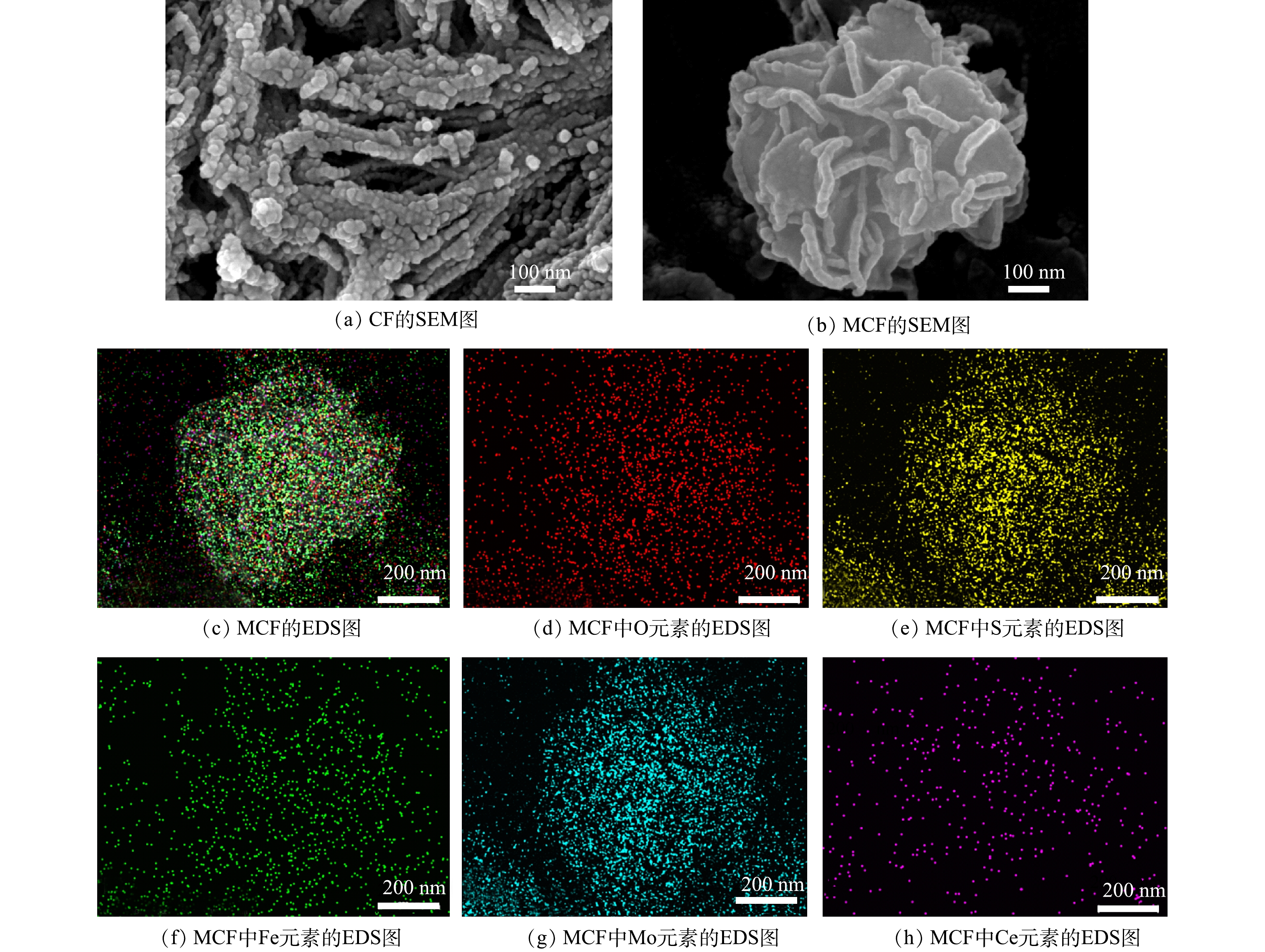
1) SEM分析。图2为CF和MCF的SEM表征图像和MCF中各元素的EDS映射图。由图2(a)可以看出,CF是由许多纳米小球组合链接并形成类似束状的物质。由图2(b)可以看出,MoS2是由厚度为10 nm左右的不规则纳米片组合而成的直径为1 μm左右的团簇花球[15]。MCF的EDS元素映射图像(图3(c)~图3(h))显示MCF中含有Fe、Ce、Mo、S、O 5种元素,说明CF较为均匀地分散附着在了MoS2花球的表面和缝隙中。因此,SEM图像与EDS元素映射图初步表明二次水热法成功合成了二硫化钼-磁性铈铁氧化物。
2) TEM分析。图3为CF和MCF的TEM图像。由图3(a)可以看出,CF的组成物质呈现出较规则均匀的颗粒状,其金属氧化物颗粒的直径为5~10 nm,这与SEM图像结果一致。图3(b)和图3(c)中,0.31 nm的晶格条纹间可以与立方萤石结构的CeO2的(111)晶面反射很好地匹配[16];图3(b)中0.19 nm的晶格条纹间距归属于斜方晶系结构的CeFeO3的(221)晶面;图3(c)中0.27 nm和0.25 nm的晶格间距可以与六方相结构的Fe2O3的(104)和(110) 2个晶面对应[17-18]。图3(d)为高清放大倍数下的MoS2边缘,由图3(d)可以清楚地识别出0.62 nm晶格间距,其对应的是六方相MoS2的(002)晶面[19]。由图3(e)可以看出,CF附着于MoS2表面。以上结果进一步说明,MCF复合材料已成功制备;并且MoS2的花球结构起到了分散CF纳米颗粒的作用,不但能降低其团聚度,同时增大了CF颗粒与PMS接触面积。
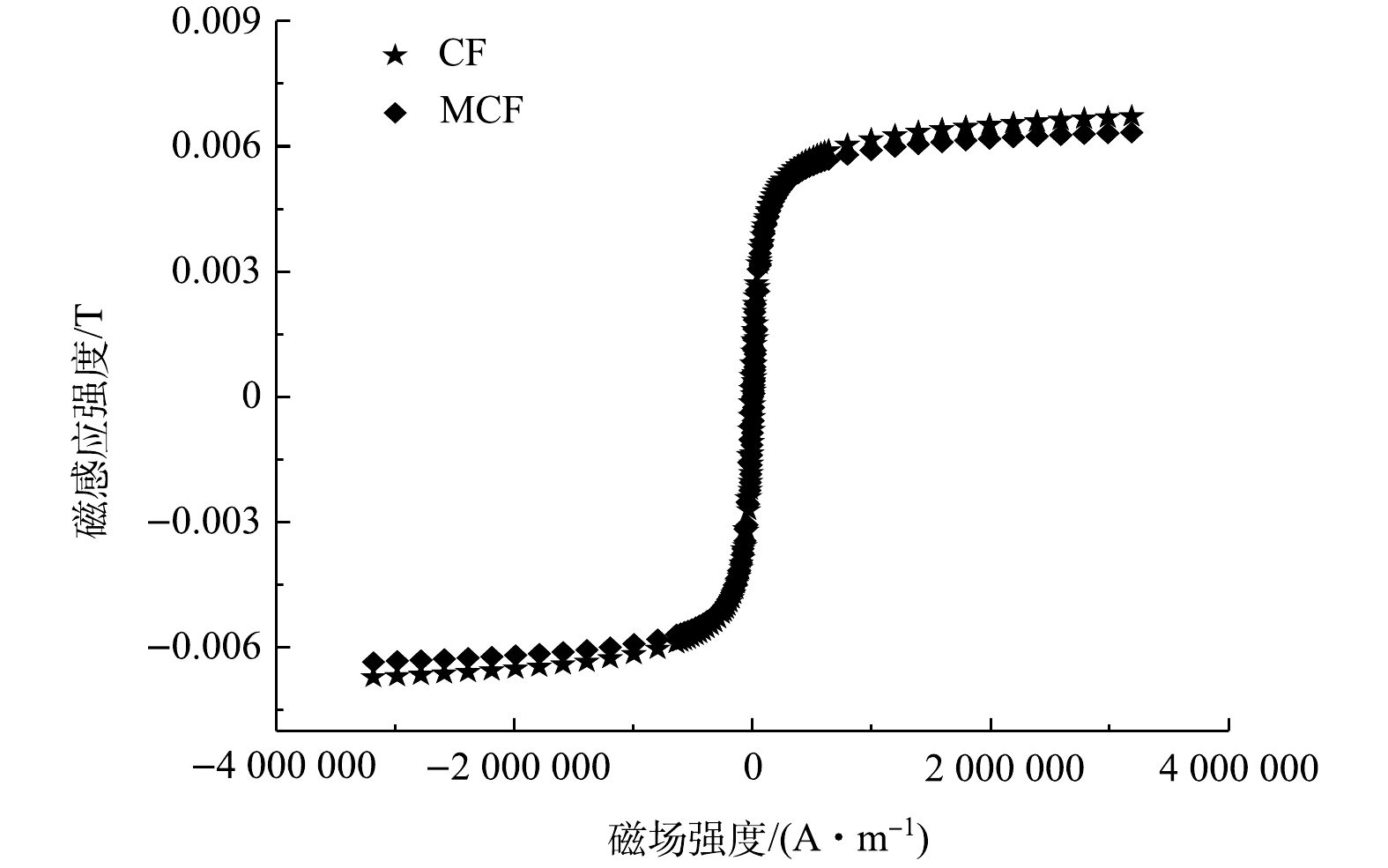
3)磁滞曲线分析。图4为CF和MCF的样品在室温下的磁化曲线。由图4可看出,CF和MCF的磁化曲线在外加磁场下均表现出典型的S型,证明他们均为磁性材料。CF和MCF的最大磁感应强度分别为6.7×10−3 T和6.3×10−3 T,MCF的最大磁感应强度略低。这主要是由于在合成MCF的过程中产生了没有磁性的MoS2,单位质量的MCF中含有的磁性物质会比CF中的少,进而导致MCF磁性下降。但MCF仍能通过外加磁力从溶液中快速分离,这对材料的回收再利用具有重要意义。
-
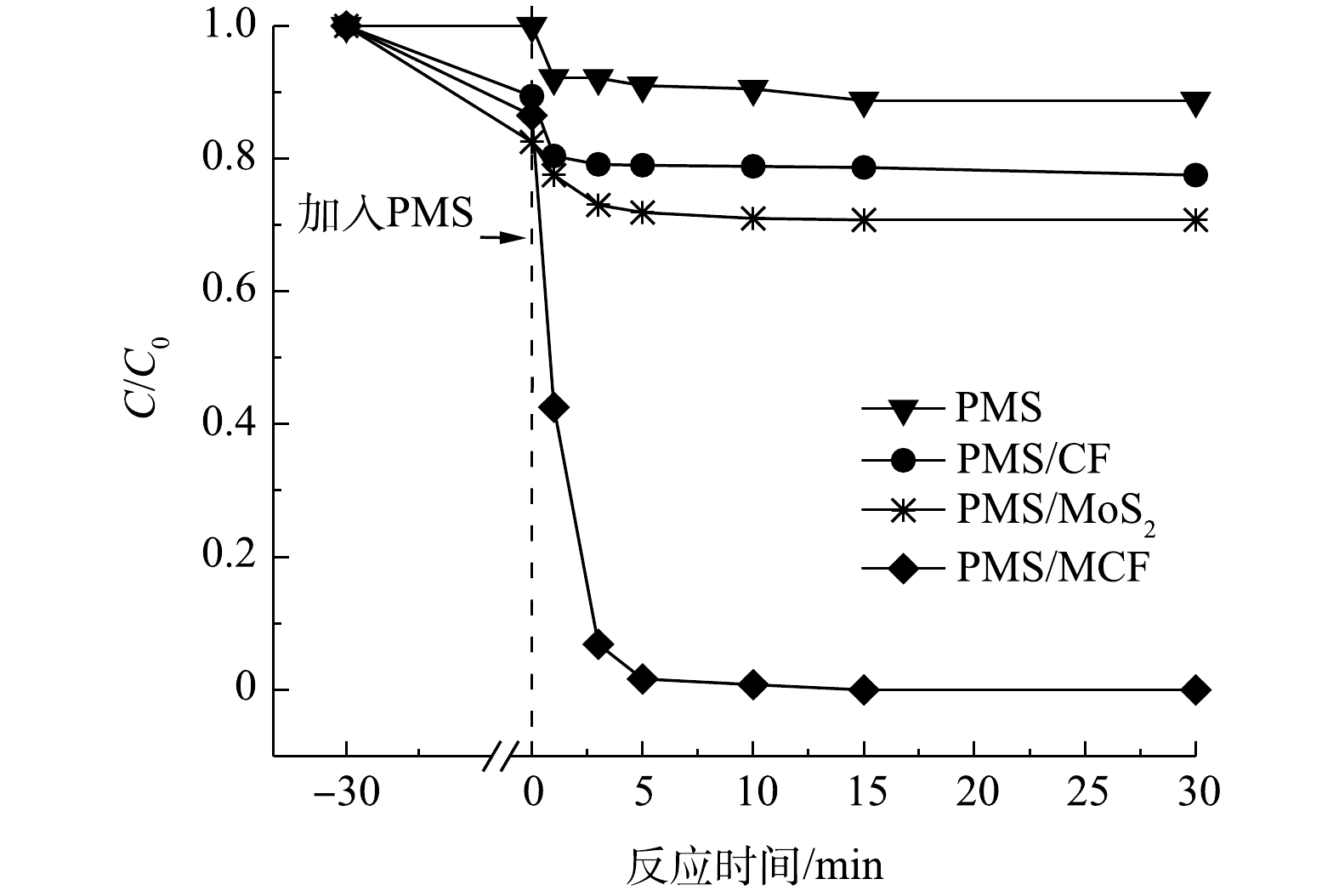
图5为不同反应体系中AO7降解率的变化。单独投加CF、MoS2和MCF均可使橙黄II的浓度有一定程度的下降,这是由于材料对AO7的吸附导致的,上述3种材料对AO7的吸附能力大小为MoS2>MCF>CF。根据CF、MoS2和MCF的SEM表征结果可知,花球状的MoS2表面褶皱较多,比表面积较大且吸附点位较多,而CF表面呈现为较为光滑的球状,导致其吸附点位较少,吸附能力弱,MCF中的CF则可能覆盖MoS2原有的吸附位点而导致其吸附能力小于MoS2[20]。在催化氧化能力方面,单独PMS、CF/PMS和MoS2/PMS体系对AO7的最终去除率分别仅有11%、22%和30%,而MCF/PMS体系对AO7的最终去除率可达到100%,说明溶液中单独的PMS氧化降解AO7的能力很弱,CF和MoS2虽对PMS具有一定的活化效果,但其活化效果远不如MCF。
-
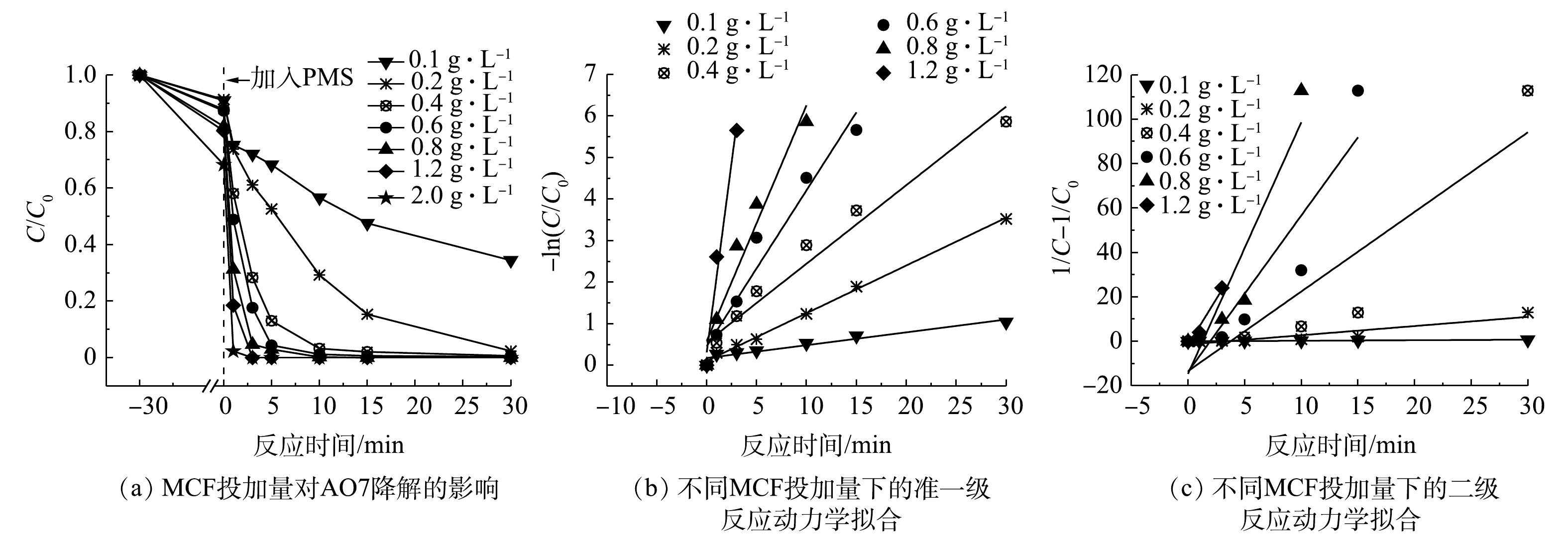
MCF投加量对AO7降解效果的影响及动力学拟合结果见图6、表1和表2。由图6(a)可看出,当MCF的投加量由0.1 g·L−1增加至2 g·L−1时,MCF对AO7的吸附能力和降解速率依次增加。这主要是由于催化剂投加量的增加使得MCF的吸附点位增多,致使AO7的吸附去除量增加;氧化去除速率的提高主要是因为较高的MCF投加量提供了更多的活化位点,能够催化PMS产生更多瞬时自由基,进而加快了AO7的降解[21]。而当MCF投加量为2 g·L−1时,降解速率并未较1.2 g·L−1时有明显提高,说明此时MCF表面激活PMS的活性点位接近于饱和,材料发生团聚,导致去除速率增幅不大。由图6(b)、图6(c)和表1、表2中的动力学拟合数据结果可知,准一级反应动力学拟合结果中的可决系数R2更接近于1,说明MCF/PMS降解AO7的过程更符合准一级动力学模型。
-
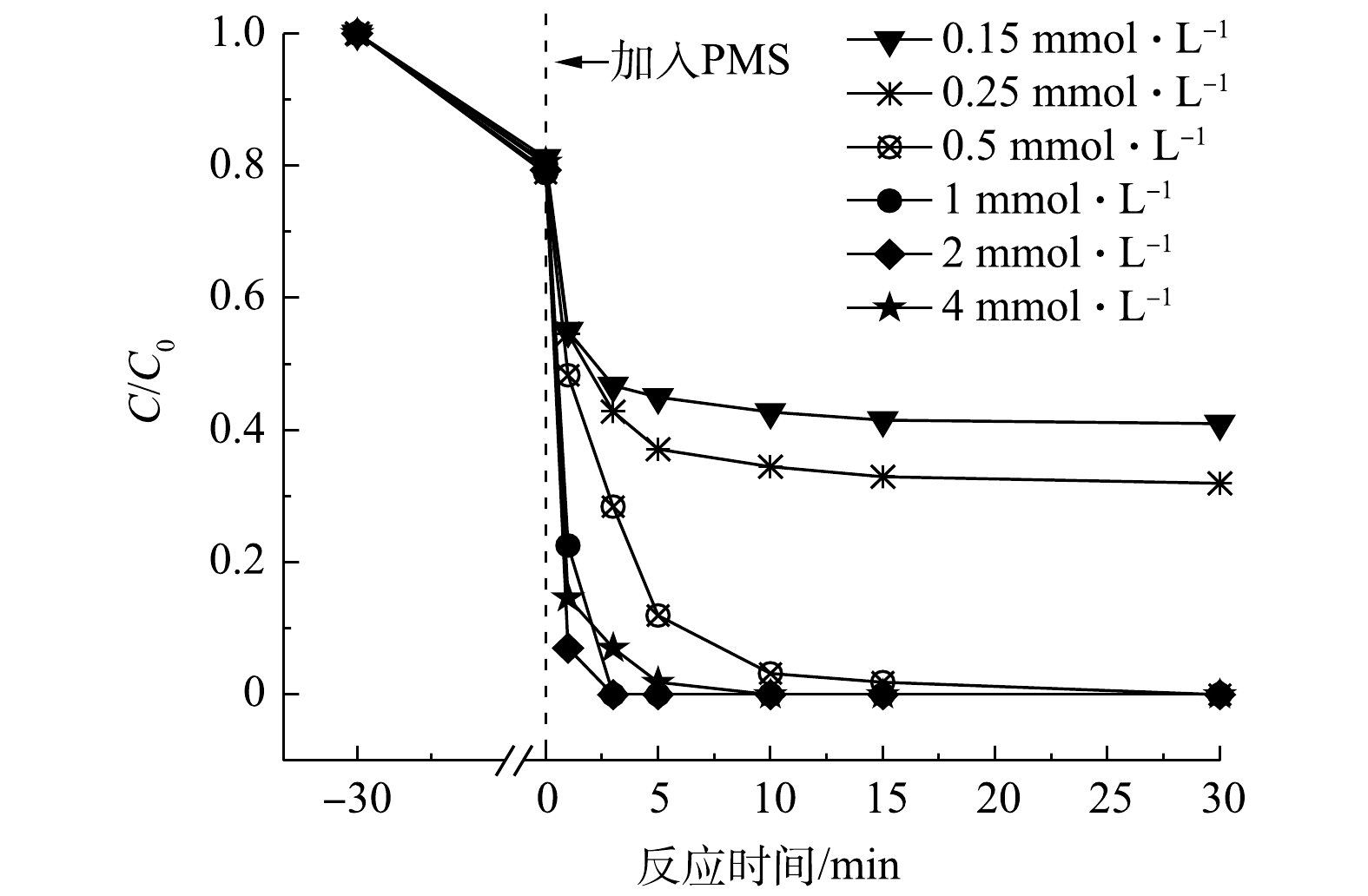
由图7可知,当氧化剂PMS的浓度低于2 mmol·L−1时,随着PMS浓度增大,AO7的降解速率随之增大。这是由于较高浓度的PMS增加了MCF与氧化剂之间的反应概率,有利于产生更多活性物种,同时促进非自由基反应[22]。而当PMS浓度为4 mmol·L−1时,降解速率明显变慢,说明过高的PMS浓度可能对氧化降解污染物的过程产生抑制作用。在满足MCF的活化点位所需的PMS含量后,体系中过多的PMS反而会消耗部分
${\rm{SO}}_4^ - \cdot $ 和·OH,生成氧化能力较弱或者不具有氧化能力的物质,导致反应速率下降[23],反应过程见式(3)~式(6)。 -
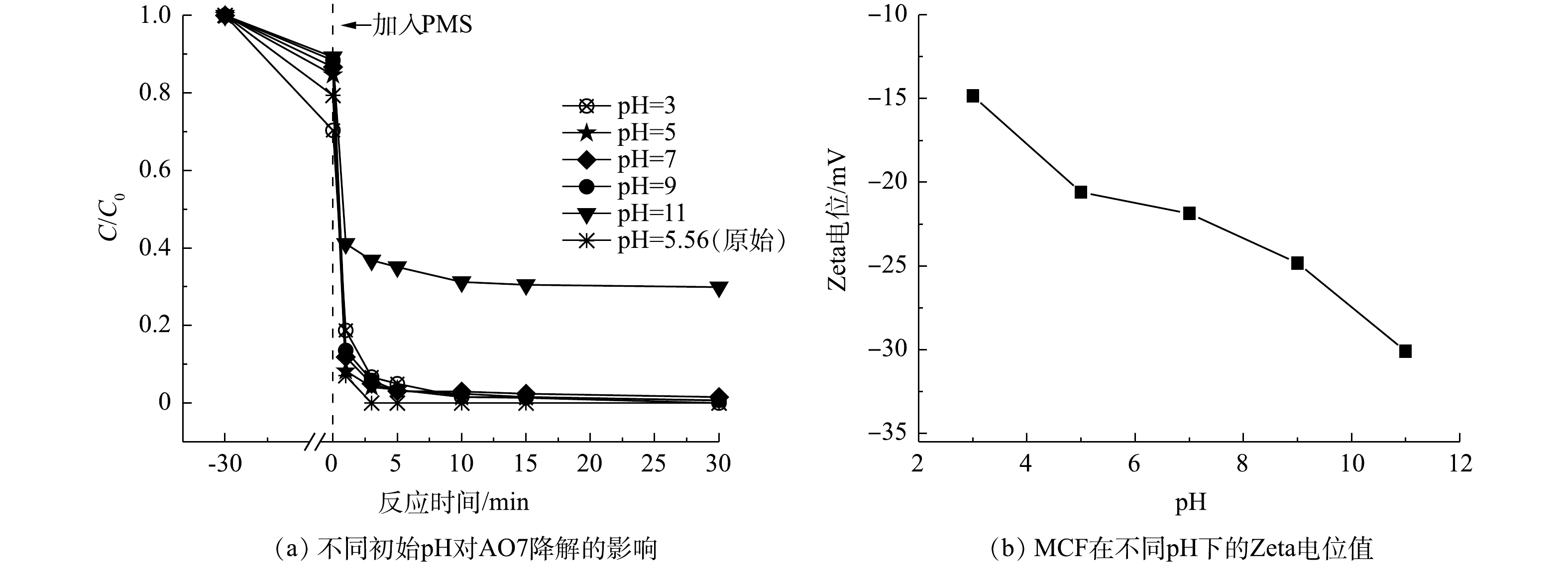
由图8(a)和图8(b)可知,MCF对AO7的吸附能力随着pH的增加而降低。在pH为3、5、7、9和11的条件下,测得MCF的Zeta电位均为负值,且随着环境pH的增高,MCF材料表面带有的负电荷离子也增多,从而增加了阴离子染料AO7与MCF间的静电斥力,进而导致MCF吸附能力的减弱[24]。由图8(a)可知,在pH为3~9时,MCF/PMS体系对AO7的最终去除率均可达到100%;而当pH为11时,MCF/PMS体系对AO7的最终去除率仅为65%。这是由于当溶液的pH过高时,氧化体系中大量的
${\rm{SO}}_4^ - \cdot $ 被转化为·OH(式(7)),但·OH的氧化能力远弱于${\rm{SO}}_4^ - \cdot $ ,从而导致体系降解污染物的能力被削弱。 -
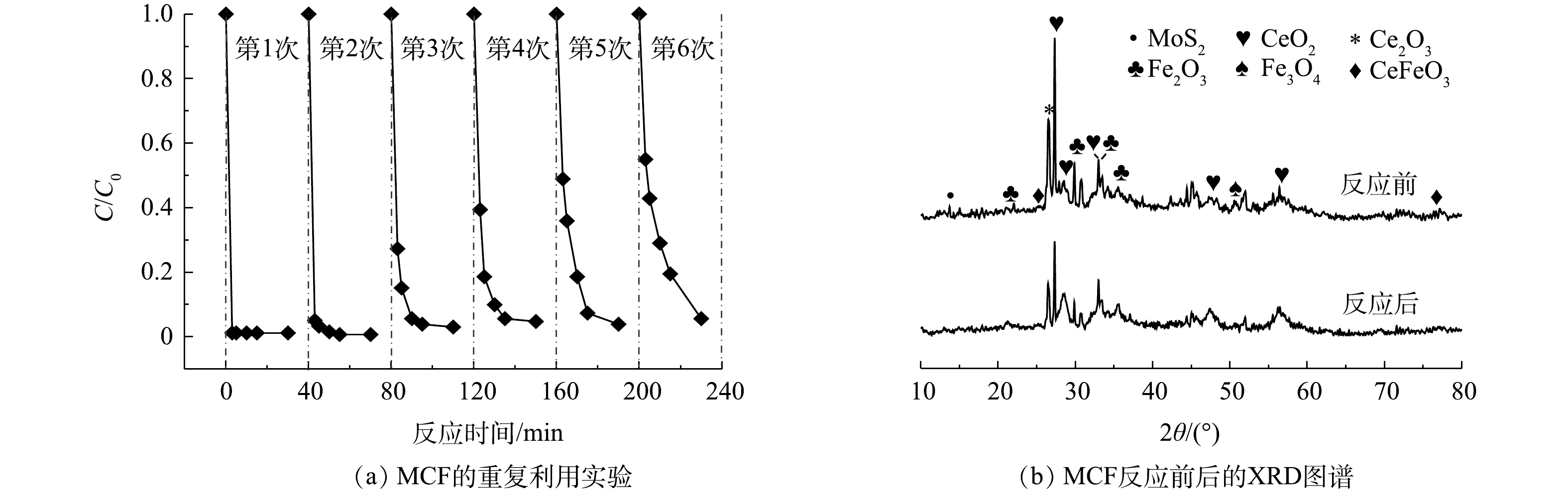
通过6次重复利用实验和对反应前后材料的XRD表征,分析了催化剂的稳定性能。由图9(a)可知,随着循环反应次数的增加,MCF/PMS体系对AO7的去除速率逐渐降低,但是经过6次循环利用后,AO7的去除率仍可达到95%,并且MCF仍可通过磁铁吸引的手段从溶液中分离出来。此结果证明,MCF具有较好的结构稳定性和可回收利用性。
由图9(b)可知,反应后的XRD谱图形状与反应前基本一致,并未出现新的衍射峰。在2θ为14.38°处出现的归属于MoS2的(002)晶面(JCPDS 37-1492)[25]的特征衍射峰强度略有降低,这可能是由于材料回收过程中MoS2少量流失所致。在2θ为28.55°、33.08°、47.48°、56.33°、59.09°和69.40°处出现的CeO2(JCPDS 34-0394,空间群为Fm-3m)[26]与2θ为27.33°处出现的Ce2O3(JCPDS 23-1048)[27]特征衍射峰依然明显,2θ为33.15°和35.61°处归属于Fe2O3(JCPDS 33-0664)的特征衍射峰[28]仍然存在,2θ为50.98°处的Fe3O4(JCPDS 28-0491)特征衍射峰[29]也没有减弱。除此之外,2θ为25.40°、47.67°和77.03°处CeFeO3(JCPDS 22-0166)的特征衍射峰[30]的强度没有发生明显变化,表明MCF提供的磁性物质并没有减少。同时,在采用ICP-OES对重复利用去除后反应溶液中的铁和铈离子浓度进行测定时发现,溶液中铁离子的溶出质量浓度低于检测限,而铈离子的最大溶出质量浓度为6.92 mg·L−1。以上结果均可说明,MCF在活化PMS降解AO7的反应后分子结构没有明显变化,具有较好的稳定性。
-
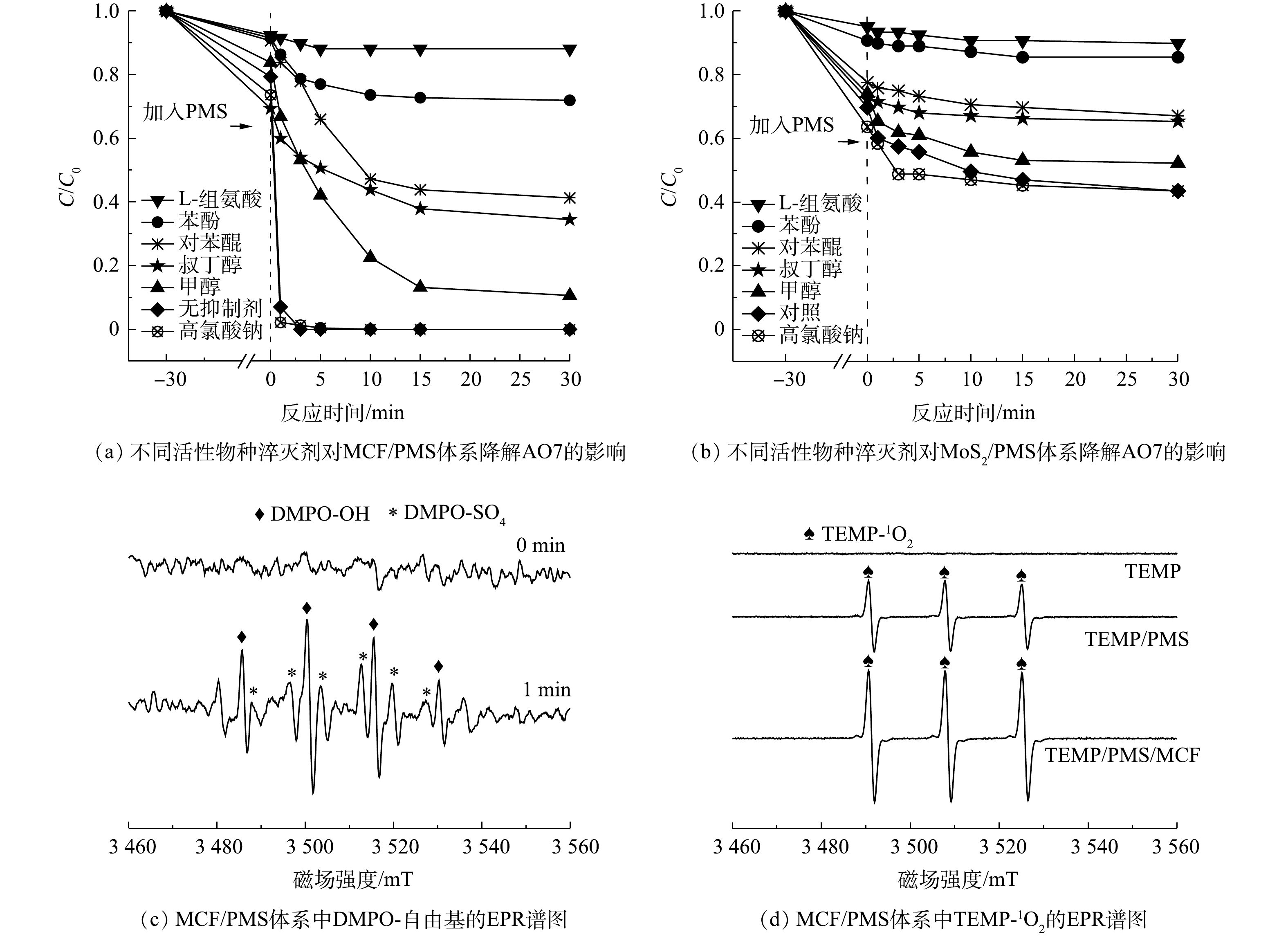
1)活性物种淬灭实验。图10为MCF和MoS2活化PMS的体系中不同功能活性物种抑制剂对AO7降解的影响和MCF/PMS体系中的活性物种检测EPR谱图。如图10(a)所示,在不添加任何抑制剂的情况下,AO7最终去除率可以达到99%以上。而向体系中分别加入浓度为2 mol·L−1的甲醇、叔丁醇和苯酚时,AO7的最终去除率分别被降低至90%、66%和28%。这说明反应中产生了
${\rm{SO}}_4^ - \cdot $ 和·OH,且抑制剂对降解AO7的抑制率排序为苯酚>叔丁醇>甲醇,这与其他金属离子活化PMS降解污染物得出的结果一致[31]。叔丁醇可抑制溶液中的·OH,苯酚和甲醇均可作为${\rm{SO}}_4^ - \cdot $ 和·OH的抑制剂,但苯酚的抑制效果最强,这主要是因为甲醇和叔丁醇不会对${\rm{HSO}}_5^ - $ 与催化剂表面的接触造成阻碍,而更多的是与溶液中存在的${\rm{SO}}_4^ - \cdot $ 和·OH发生反应。但污染物却仍有可能在接近MCF表面时被自由基氧化[32],而苯酚则能够同时抑制材料表面及附近的自由基产生,因此,苯酚呈现出更强的抑制效果。与不添加抑制剂的反应体系相比,加入L-组氨酸、对苯醌和高氯酸钠的反应体系对AO7的最终去除率分别为12%、59%和100%。这说明L-组氨酸和对苯醌分别对反应有不同程度的抑制作用,即1O2和
$ \cdot {\rm{O}}_2^ - $ 也作为重要的活性物质参与到了氧化反应中。而高氯酸钠不仅没有抑制降解过程,反而对AO7的去除速率有轻微的促进作用,证明在本实验中并不存在其他的非自由基反应机制。根据图10(b)可知,与上述相似的抑制趋势可以在MoS2/PMS体系中看到,再次说明MoS2也具有一定的活化PMS产生活性物种的能力。使用DMPO作为自旋阱捕获MCF活化PMS产生的·OH和
${\rm{SO}}_4^ - \cdot $ 的结果如图10(c)所示,在加入MCF之前(0 min)并没有出现自由基的峰值,即单独的DMPO几乎不产生自由基。而在加入MCF后1 min的EPR波谱中,可以明显看到DMPO-SO4和DMPO-OH加合物的强峰,这再次证明MCF/PMS体系中产生了${\rm{SO}}_4^ - \cdot $ 和·OH,并且DMPO-OH的信号明显高于DMPO-SO4。这与添加叔丁醇和甲醇的淬灭实验结果一致,即表面上看来·OH的作用似乎比${\rm{SO}}_4^ - \cdot $ 更大,原因可能是体系中的${\rm{SO}}_4^ - \cdot $ 会快速与H2O发生不理想的亲核取代反应并转化为·OH[33](图11)。因此,与·OH相比,${\rm{SO}}_4^ - \cdot $ 在氧化降解AO7的过程中做出了更大贡献。此外,由图10(d)可知,在单独添加TEMP的体系中未观察到特征信号,说明自旋捕获剂TEMP对整个反应体系没有影响。但在PMS/TEMP体系中产生了相对较强的TEMP-1O2加合物1∶1∶1的三重态特征信号。这是由于水溶液中的PMS可以通过自行分解产生少量的1O2[34]。而添加MCF后,TEMP-1O2加合物强度显著增强,根据以往的研究,非均相催化反应中的1O2一般来自碱/苯醌活化[35]和PMS的自分解。而本实验中较高pH条件下和添加苯醌的体系中降解反应均受到抑制,说明MCF对PMS的自分解起到了很强的促进作用。综上所述,在MCF/PMS降解AO7体系中产生的1O2、
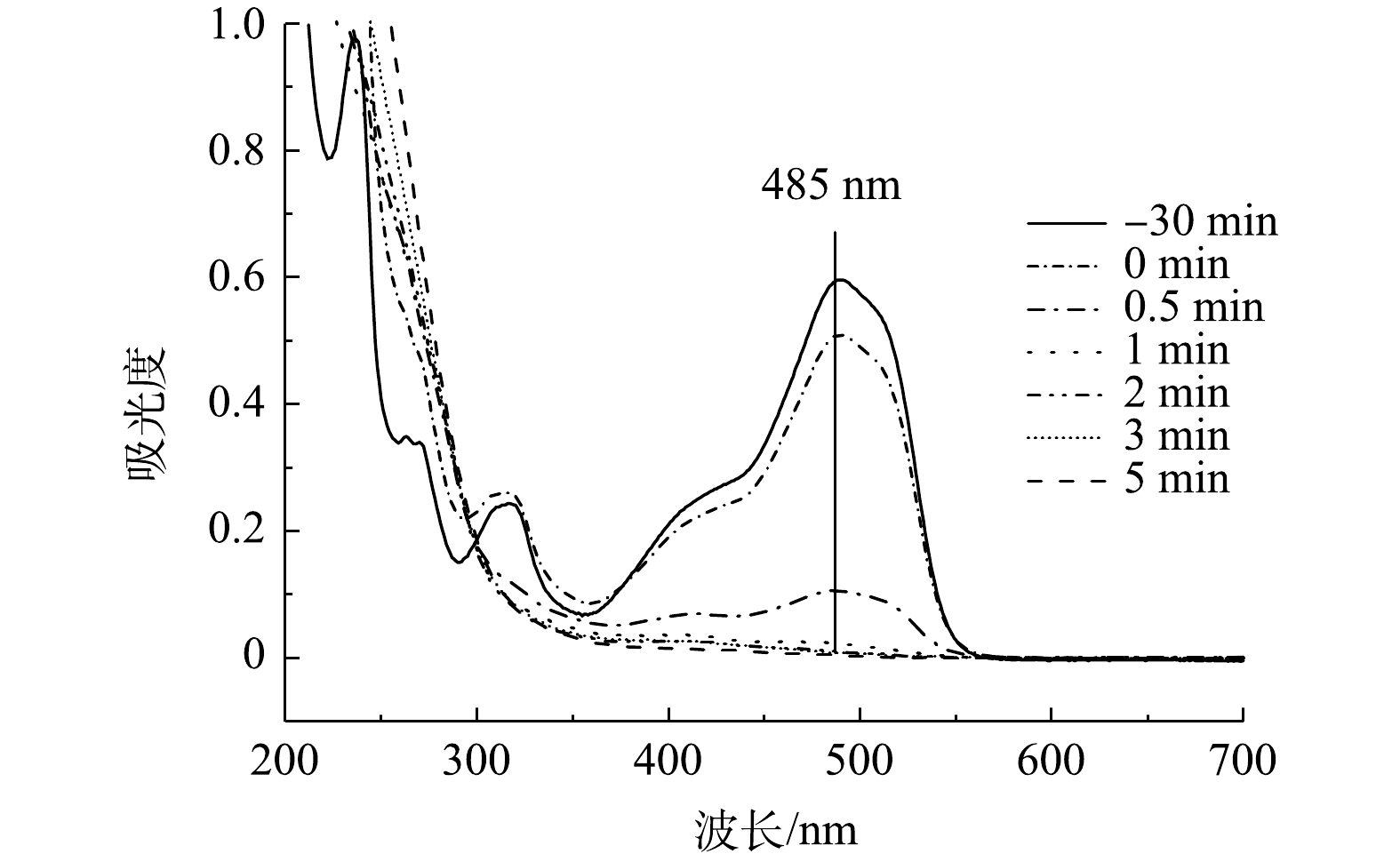
${\rm{SO}}_4^ - \cdot $ 、·OH、$ \cdot {\rm{O}}_2^ - $ 均对氧化过程有一定的贡献,1O2、${\rm{SO}}_4^ - \cdot $ 和·OH在降解反应中起主导作用。2)降解UV-vis图谱分析。由图12可知,AO7的特征波长分别为485、310和228 nm,上述3个波长处的吸收峰分别对应其偶氮发色基团(—N=N—)、萘环和苯环结构[33, 36]。随着反应时间的增加,可以看到485 nm处对应的吸收峰强度不断降低至几乎为0,说明溶液中所有AO7分子的—N=N—均发生了断裂,这与图12中的脱色结果一致;310 nm处的吸收峰在反应过程中逐渐消失,而228 nm处的吸收峰在反应后期变强,说明AO7分子中的—N=N—断裂后形成了一些萘环和苯环芳香碎片,并且萘环碎片有被氧化分解的现象[37]。
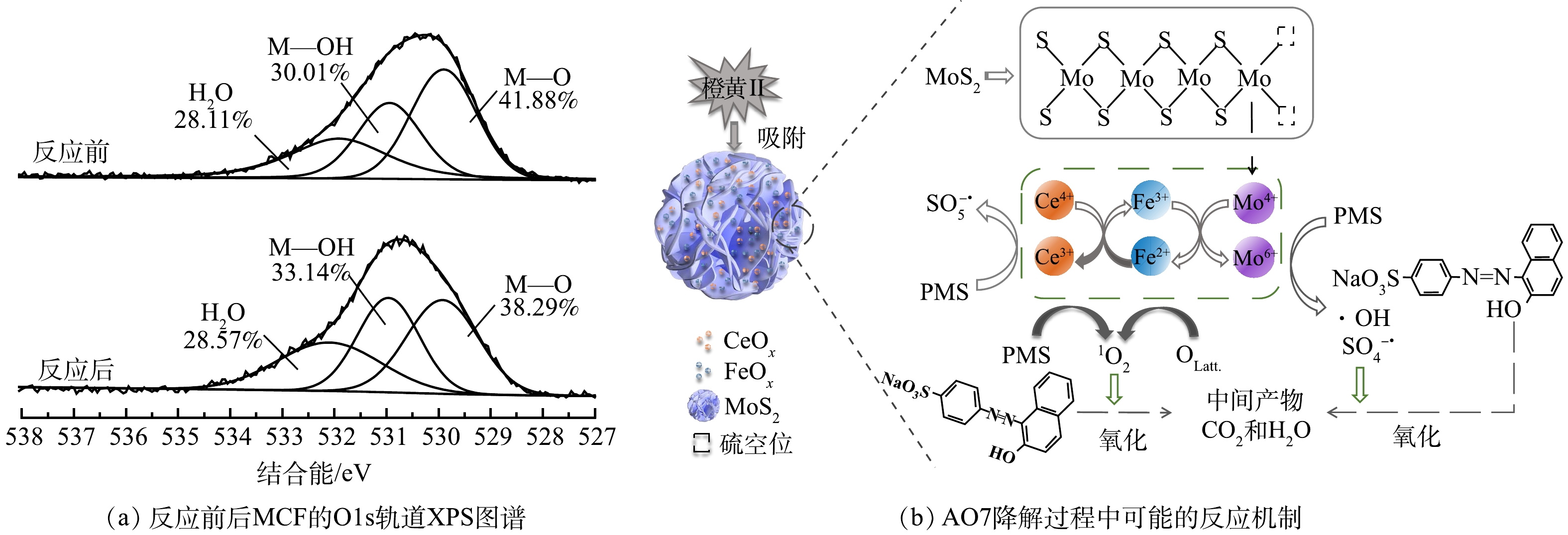
3)反应机理初步推导。前述分析结果表明,MCF活化PMS的过程中产生了1O2、
${\rm{SO}}_4^ - \cdot $ 、·OH和$ \cdot {\rm{O}}_2^ - $ 几种活性物种,其中1O2主要由PMS的自分解产生。此外,由图13(a)可知,反应后MCF的晶格氧(M-O)含量由41.88%下降为38.29%,说明材料中的晶格氧也参与了1O2的生成。活性物种${\rm{SO}}_4^ - \cdot $ 和·OH主要由体系中电荷转移所产生。图13(b)反映了AO7降解过程中可能的反应机制。在降解过程中,有较少部分AO7是通过PMS直接氧化和MCF的吸附作用去除的,MoS2的花球状结构有利于污染物的吸附与传质,剩余绝大部分AO7则是通过MCF/PMS体系的氧化降解作用而被去除。淬灭实验结果说明,在MoS2/PMS和MCF/PMS体系中起到主导作用的均是1O2。在MCF活化PMS的过程中,MoS2具有较大的比表面积和较高的电子转移速率,MCF中CF和MoS2的关联作用有利于PMS活化过程中的电荷转移,从而加速了
${\rm{SO}}_4^ - \cdot $ 和·OH的生成,即Fe2+、Fe3+、Ce3+、Ce4+和Mo4+可以通过电荷转移活化PMS产生${\rm{SO}}_4^ - \cdot $ 和·OH(式(8)~式(12)),而MoS2表面的硫空位和暴露出的还原金属活动中心Mo4+还可以通过提高界面电荷转移效率起到增强Fe3+/Fe2+氧化还原循环的作用(式(13))[38]。在体系未添加MCF时,PMS自分解可产生小部分的1O2(式(14)和式(15)),体系中添加的MCF促进了PMS的自分解产生大部分1O2,并且还有一部分1O2由MCF中的晶格氧(OLatt.)转化得到(式(16)和式(17));此外,低价金属离子在反应过程中均起到了活化PMS产生1O2的作用(式(18)和式(19))。Ce4+/Ce3+和Fe2+/Fe3+的标准氧化还原电位分别为1.44 V和0.77 V,因此,电子从Fe2+转移到Ce4+的过程从热力学角度来看也是实际可行的[8](式(20))。综上所述,体系中产生的1O2、${\rm{SO}}_4^ - \cdot $ 和·OH可将AO7降解为中间产物,并可进一步将中间产物完全矿化为CO2和H2O(式(21))。
2.1. 催化剂表征分析
2.2. 不同体系AO7降解效果
2.3. 催化剂投加量的影响
2.4. 氧化剂浓度的影响

2.5. 溶液初始pH的影响


2.6. 催化剂稳定性评价
2.7. 反应机理初步分析



















-
1)采用二次水热法成功制备了MCF复合材料,CF以颗粒形式附着于花球状MoS2的表面。
2) MCF呈现比CF和MoS2更显著的催化活性,MCF中的CF和MoS2在反应过程中存在协同作用。在MCF投加量为1.2 g·L−1、PMS浓度为2 mmol·L−1、pH为3~9时,MCF/PMS体系对AO7的去除率在反应3 min即可达到100%。
3) MCF的化学性质稳定,且有一定的可回收利用性。在AO7降解过程中起主导作用的活性物种为1O2、
${\rm{SO}}_4^ - \cdot $ 和·OH,1O2来自于PMS自分解和晶格氧的转化,${\rm{SO}}_4^ - \cdot $ 和·OH由金属离子活化PMS产生。AO7最终被降解为含有萘环和苯环的中间产物、CO2和H2O。


