

 下载:
下载:




-
近年来,随着全球工业化的不断发展,化石能源的使用排放出大量温室气体(CO2、CH4、N2O、O3和氯氟烃等)。自1800年至2020年,大气中CO2的体积浓度由280 mL·m−3升至410 mL·m−3,已导致全球气温上升约1.2 ℃[1-2]。全球变暖将对生态环境以及人类生存造成显著威胁[3-4]。2020年9月22日,中国在第七十五届联合国大会提出“二氧化碳排放力争于2030年前达到峰值,努力争取2060年前实现碳中和”的目标[5]。为实现此目标,有效缓解全球变暖,应在大力开展新能源研究和应用的同时,研发经济高效的CO2捕集、利用与封存技术(carbon dioxide capture utilization and storage, CCUS)[6]。
矿物碳酸化封存CO2技术凭借矿石资源丰富、碳酸盐产物稳定无污染、操作简单等优点,被认为是除胺法等溶剂吸收法和地质封存的可行性CO2捕集方案[7]。但天然矿石封存CO2会消耗大规模矿产资源,而工业固体废物(粉煤灰、钢渣、电石渣等)通常含有大量的钙、镁元素,可作为碳酸化所需钙离子、镁离子来源以替代天然矿石对CO2进行捕集和封存[8-9]。任国宏等[10]运用直接湿法研究了粉煤灰、电石渣及其配合物的矿化封存CO2效果,在60 ℃、1个标准大气压(1.01×105 Pa)条件下,粉煤灰、电石渣固碳率分别是2%和61.3%,其配合物固碳率比等量单一电石渣和粉煤灰固碳率之和计算值提高19.6%。伊元荣等[11]研究了钢渣湿法捕获CO2的反应特性,发现钢渣中的Ca(OH)2、CaO、Ca2SiO4和Ca3AlO6等矿物都可以发生碳酸化反应生成CaCO3,并论证了矿化封存CO2后的钢渣可被进一步利用为建材。碳酸化反应后的灰渣可用于各种行业,包括建筑材料[12-14]、纸张和涂料填料、耐火材料、农业和制药[15],其主要利用行业是建筑业。据估计,通过利用碳酸化产品生产建筑材料来替代传统的碳密集型产品,可间接减少约3.7×1010 t的二氧化碳排放[16]。除了用于生产经济价值较低的常规建筑材料,通过矿化过程回收附加值较高的产品是未来提高市场竞争力的一大方向[17-19]。
国内外学者对不同固废封存CO2进行了大量基础性研究,并探索了直接湿法和干法等工艺的封存应用效果[20-23]。固废的化学成分及物相组成差异对CO2封存能力影响巨大[24],而文献中对同种固废分别采用2种工艺进行CO2封存能力及规律性的对比研究仍十分有限。本课题组选择产量大、源自不同行业(气化、冶炼、化工)的3种典型工业固体废物(电石渣、钢渣和粉煤灰),分别通过湿法高压釜间歇装置和固定床干法连续装置,对比研究直接湿法和直接干法工艺下其CO2封存能力及规律,并结合分析表征,揭示不同工艺下固废碳酸化反应机理,以期为进一步开发低成本绿色高效封存CO2技术提供参考。
全文HTML
-
实验所用工业固体废物有钢渣(steel slag,SS)、电石渣(carbide sludge,CS)和粉煤灰(fly ash,FA)。SS包括山东某碳钢厂钢渣(记为SS-1)和山东某不锈钢厂钢渣(记为SS-2);CS取自宁夏某煤化工厂电石渣;FA取自天津某炼化厂煤气化粗渣。表1为各固废样品的成分组成。样品首先经105 ℃干燥至恒重,然后采用120目和200目的筛子在振筛机筛分5 min,得到粒径为75~120 μm的样品,密封保存。实验所用纯CO2、N2购于北京环宇京辉气体科技有限公司,体积分数为99.9%。去离子水采用威立雅PURELAB Flex超纯水仪自制,电阻率为18.2 MΩ·cm。实验中新鲜样品分别可用F-SS-1、F-SS-2、F-CS、F-FA表示,反应后样品用C-SS-1、C-SS-2、C-CS、C-FA表示。
-
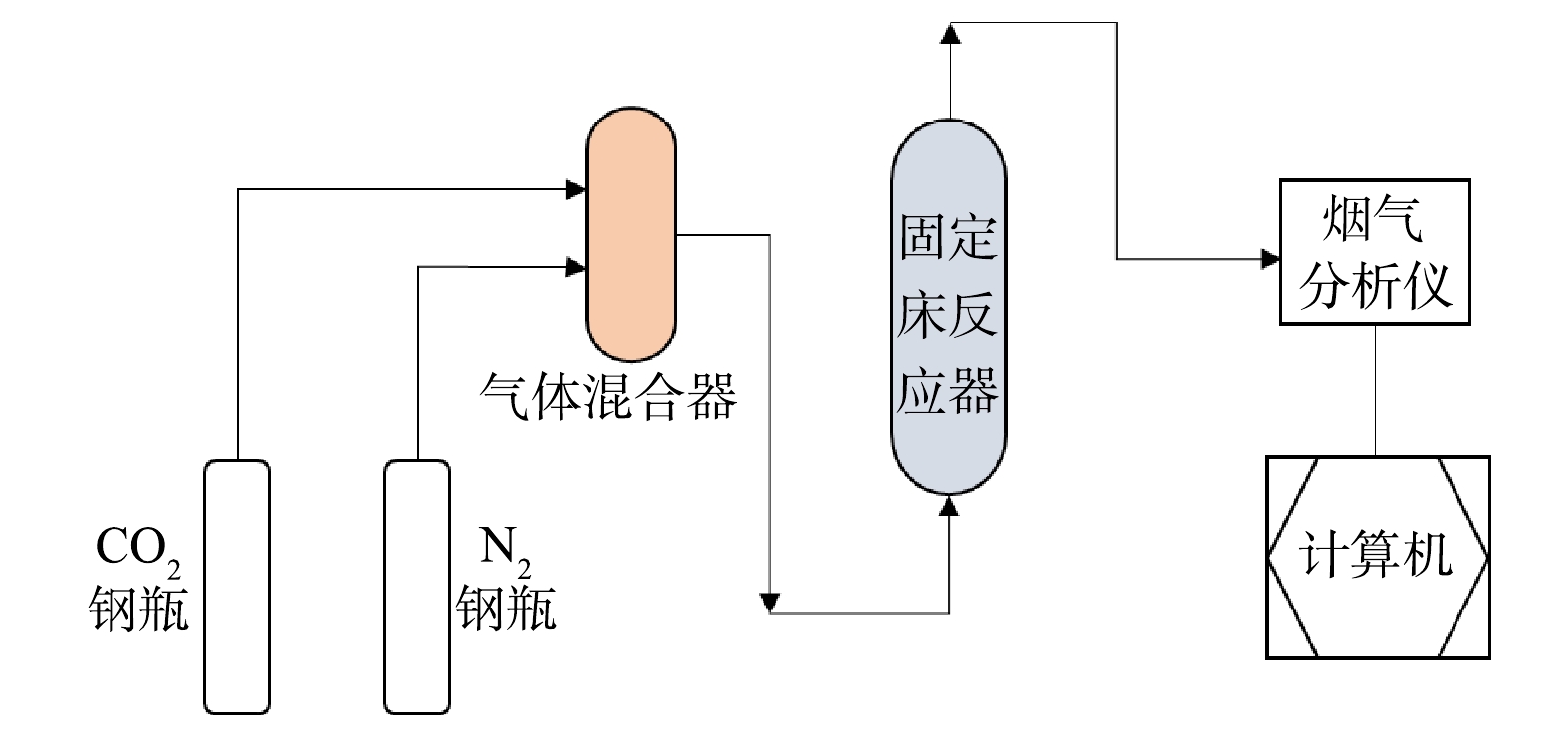
1)干法固定床连续装置(见图1)由气体混合部分(纯N2、CO2气源、气体混合罐、流量控制器)、气固反应部分(固定床反应器)及烟气分析部分(红外烟气分析仪、计算机)组成。不锈钢管式反应器内径为50 mm,长700 mm,设计温度900 ℃,设计压力4.5 MPa。采用传感器测定气体流量、反应器温度、压力等,通过数据采集系统转换成数字信号,存入电脑并处理。
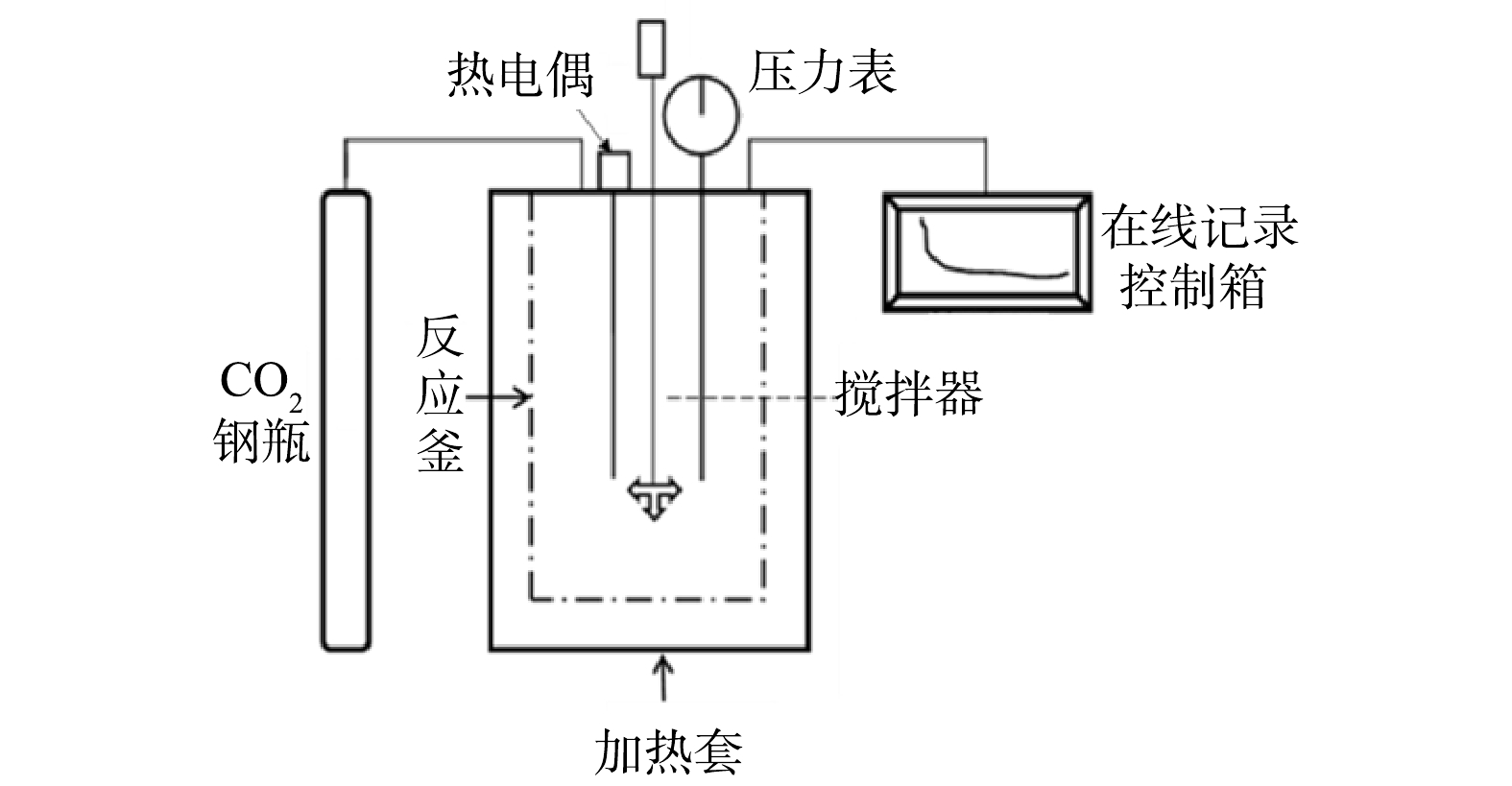
2)湿法高压釜间歇装置如图2所示。该装置主要由高压反应釜(KTFD06-20型,烟台科立化工设备有限公司产品)、纯CO2气源、在线记录控制箱3部分组成。反应釜内部体积为0.6 L,工作温度300 ℃,工作压力20 MPa,工作转速200 r·min−1。采用传感器测定反应器温度、压力、转速等,通过数据采集系统转换成数字信号,存入电脑并处理。
-
1)直接干法:将10 g经预先干燥的固废样品装填于反应器中,充入高纯度N2排空装置内空气,升温至反应温度;通过调节N2、CO2流量控制器,控制CO2体积浓度为15%,在总气体流速200 mL·min−1、常压(0.1 MPa)条件下,打开阀门将气体通入反应器,与固废进行反应;通过烟气分析仪实时记录反应器出口气体流量和CO2体积分数的变化,直到出口CO2体积分数和流量不变,反应结束。据此计算固废的CO2封存量,最后采用分析表征方法对固体产物进行分析。
2)直接湿法:将150 mL去离子水和10 g经预先干燥的固废样品加入高压釜中;充入高纯度N2排空装置内空气,关闭出口阀门;在密闭条件下升温至设定温度65 ℃;随后将高纯度CO2气体从气瓶注入反应器中,使高压釜内的压力稳定在2 MPa;开启机械搅拌,转速200 r·min−1,同时开始计时;待反应达到2 h时停止加热,使反应釜内的温度降至25 ℃;然后对反应釜内的悬浮液用0.7 μm滤纸进行真空抽滤,抽滤后的固体产物放入恒温干燥箱,105 ℃干燥12 h,并研磨均匀;采用分析表征方法对固体产物进行分析。
-
1)直接干法:CO2封存量计算公式见式(1)。
式中:
$ Q_{{\rm{CO}}_2} $ 为CO2的封存量,g·kg−1;$ M_{{\rm{CO}}_2} $ 为CO2的摩尔质量,g·mol−1;m为灰渣的质量,g;Qin为进口气体流量,L·min−1;Qout为出口气体流量,L·min−1;Cin为进气中CO2体积分数,%;Cout为出气中CO2体积分数,%。2)直接湿法:采用德国NETZSCH公司的STA 449F3型热重分析仪进行热重实验,称取一定质量的样品,在30 mL·min−1的N2气氛下进行热失重实验。测定不同碳酸化样品中的CO2质量分数w(CO2),进而得出灰渣的CO2封存量,如式(2)~(3)所示。碳化率
$ \left( {{{{\zeta }}_{{\rm{Ca}}}}} \right)$ 即碳酸化效率,表示灰渣中用于碳酸化的钙质量占总钙质量的比例,根据式(4)计算。式中:
$ \Delta {{{m}}_{{\rm{550 \sim 95}}{{\rm{0}}^ {\;\;\circ }{\rm{C}}}}} $ 为550~950 ℃的质量损失对应碳酸盐分解带来的质量损失,g;w(CO2)为碳酸化样品中的CO2质量分数;$ Q_{{\rm{CO}}_2} $ 为CO2的封存量,g·kg−1;MCa和${M_{{\rm{CO}}}}_{_2} $ 分别代表Ca和CO2的摩尔质量,g·mol−1;Catotal代表新鲜灰渣样品中Ca的质量分数。 -
采用X射线荧光光谱(X ray fluorescence, XRF)技术分析固废样品化学组成。采用Empyrean Panalytical型X射线衍射仪(X-ray diffraction, XRD)对碳酸化前后灰渣样品中矿物相进行定性分析。衍射条件为:Cu靶(λ=0.154 nm),扫描角度范围10°~70°,扫描步长为0.01°,管电压40 kV,管电流40 mA。采用Bruker公司的Tensor27型傅里叶变换红外光谱仪(FTIR)对碳酸化反应前后灰渣样品进行主要化学键分析,操作条件为:溴化钾压片,波数400~4 000 cm−1,分辨率1 cm−1。采用FEI公司的Quanta-200型场发射扫描电子显微镜(SEM)观察碳酸化反应前后灰渣样品的微观形貌。工作前,将固废样品固定在环氧树脂中,抛光,并安装在样品柱上,进行喷碳处理。操作条件为:电压10 kV,电流密度45 μA·cm−2,电子束角度90°。采用配套的能量色散谱(EDS)分析确定样品的微区化学组成。比表面积、总孔体积、吸附/脱附等温线及孔分布在Quantachrome AS-3静态氮吸附仪上测定。测定前,将样品置于样品处理系统,在300 ℃下抽真空至1.33×10−2 Pa,恒温恒压4 h,净化样品。比表面积通过Brunauer-Emmett-Teller(BET)方法计算得到。总孔体积根据相对压力约为0.99时吸附液氮总量计算得到。
1.1. 实验材料
1.2. 实验装置
1.3. 实验步骤
1.4. 数据处理






1.5. 分析与表征
-
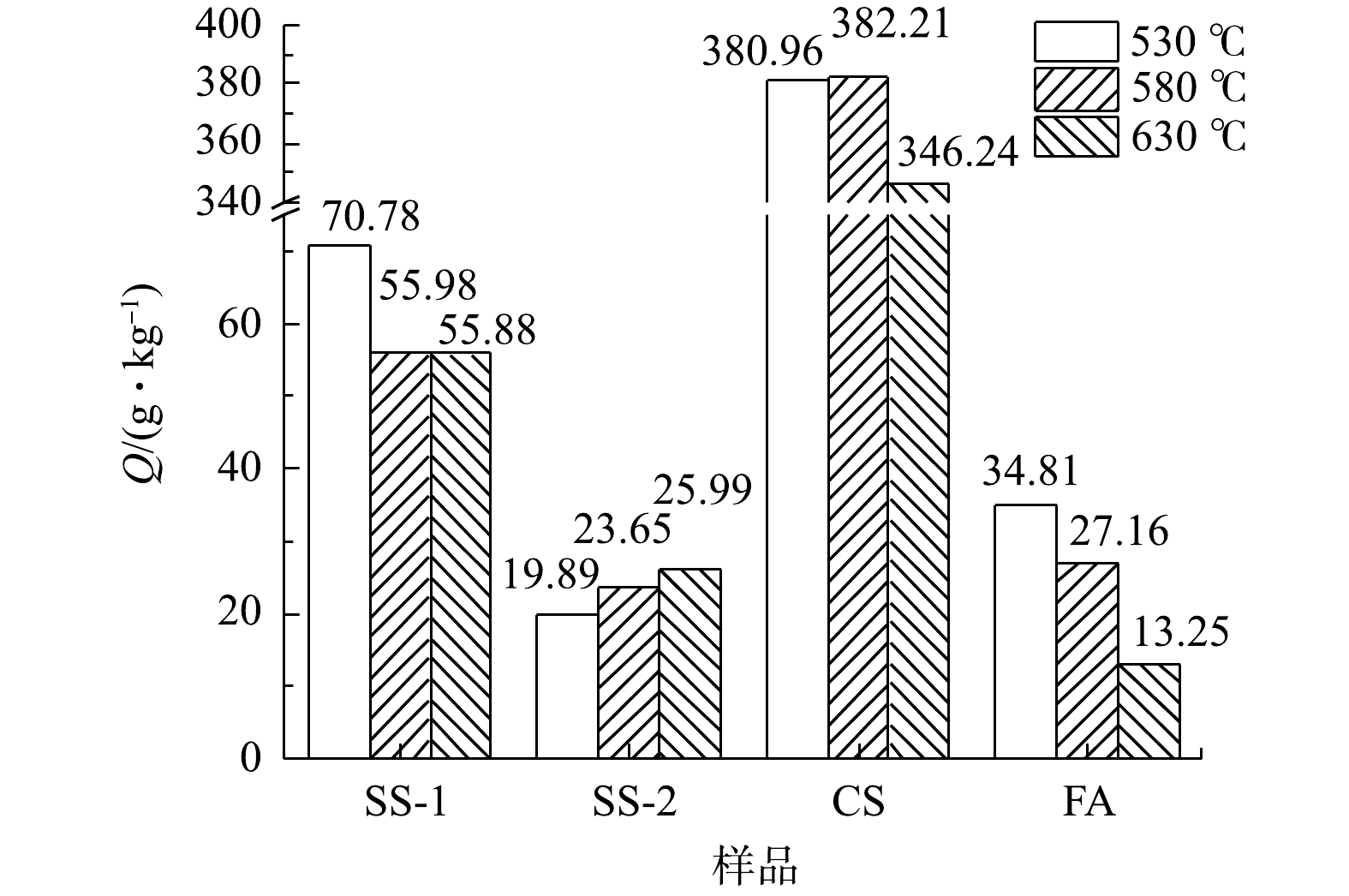
在不同温度下4个样品的CO2封存量如图3所示。由图3可知,SS-1样品随着温度升高,CO2封存量逐渐降低,但在580 ℃和630 ℃时封存量基本持平,530 ℃时取得最大封存量70.78 g·kg−1;SS-2样品随着温度升高,CO2封存量逐步升高,在630 ℃取得最大封存量25.99 g·kg−1;CS样品随着温度升高,CO2封存量先增大后减小,在580 ℃取得最大封存量382.21 g·kg−1;FA样品随着温度升高,CO2封存量降低,在530 ℃取得最大封存量34.81 g·kg−1。总体来看,CO2封存能力呈现出CS>SS-1>FA>SS-2的规律,其中,FA和SS-2的差距很小。结合各灰渣组分信息分析可发现,固废样品CO2封存能力与碱性组分CaO含量具有一定的相关性,CS、SS-1、FA 3个样品中CaO组分含量依次减小且差距较大,与其CO2封存能力呈正相关。另外,虽然SS-2的CaO含量较高,但由于FA和SS-2中的SiO2含量都比较高,两者的CO2封存能力也呈现出相应较低的水平,故SiO2的含量较大程度影响了SS-2和FA的CO2封存效果,这可能与活性钙的物相有关。因此,固废中CaO含量是影响干法矿化封存CO2方法封存效果的重要因素。
-
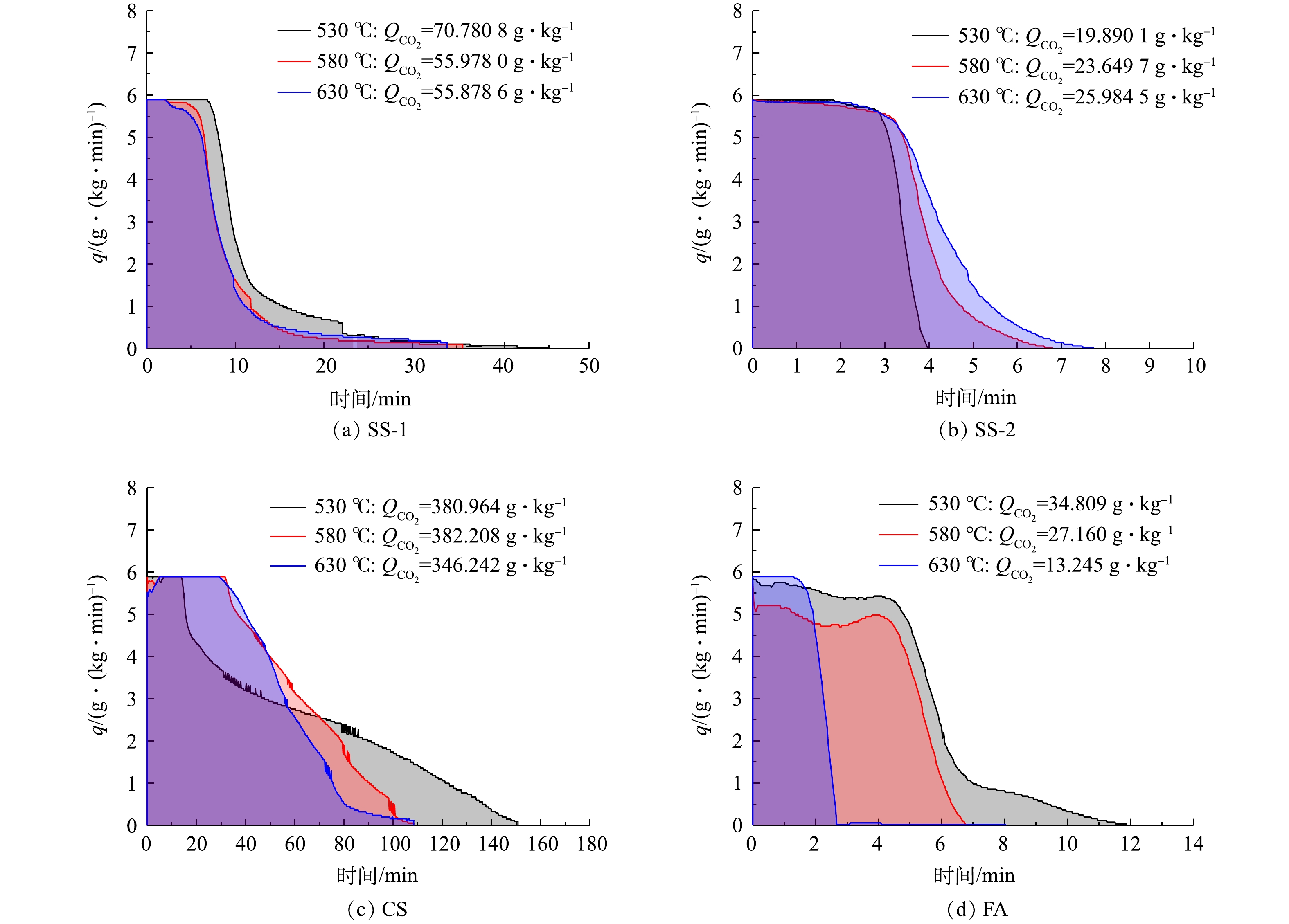
图4为各固废样品封存CO2过程中的碳酸化反应速率曲线。由图4可见,在不同温度下,各样品干法矿化封存CO2反应前期吸收速率最大,恒定一段时间,随后在某一点吸收速率瞬间开始下降。SS-1样品在40 min内基本完成反应,且在反应开始10 min后封存速率急速下降;CS样品反应时间长,需2 h左右反应完全,CO2封存速率在反应开始40 min后逐步下降;SS-2和FA在12 min内基本完成反应,封存能力低,且在2~4 min时封存速率急速下降。结合分析表征可推测,反应生成的碳酸钙覆盖在颗粒表面或发生团聚,减少了与气体反应的接触面积,且活性钙在前期反应中已消耗很多,进而影响了反应速率。因此,在CO2速率曲线上呈现出反应初期为快速化学反应控制阶段,反应后期为慢速扩散控制阶段。
-
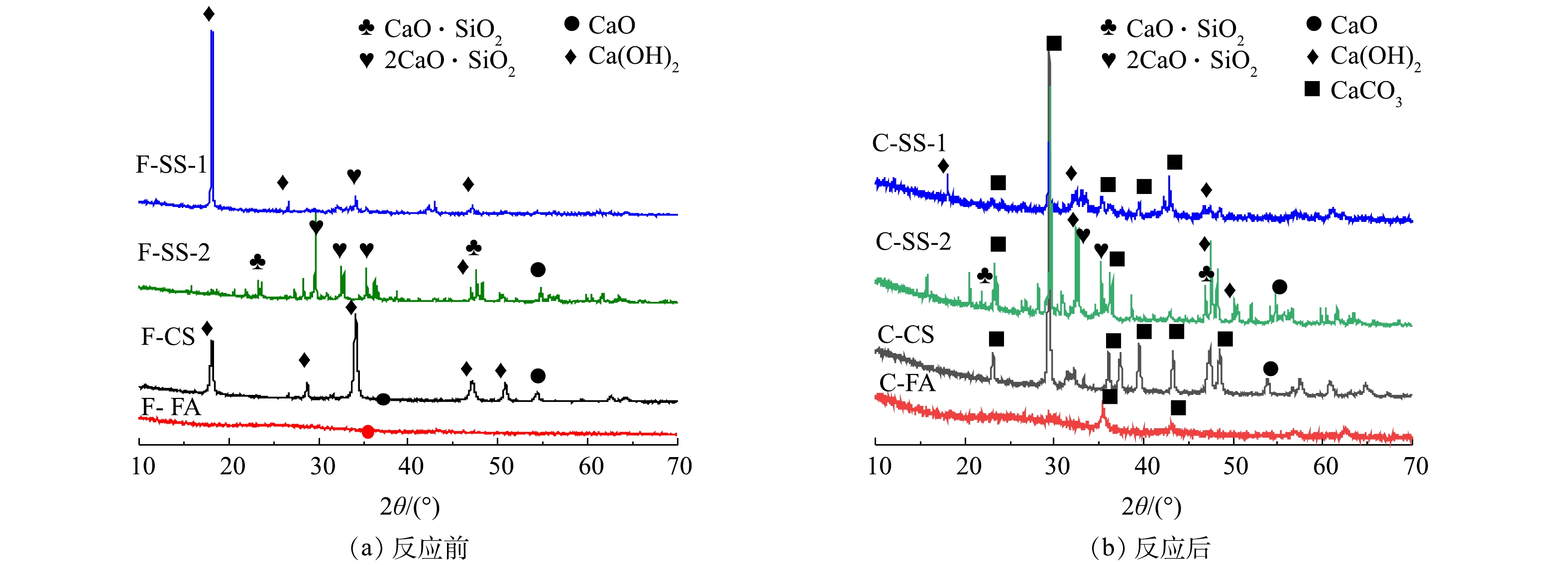
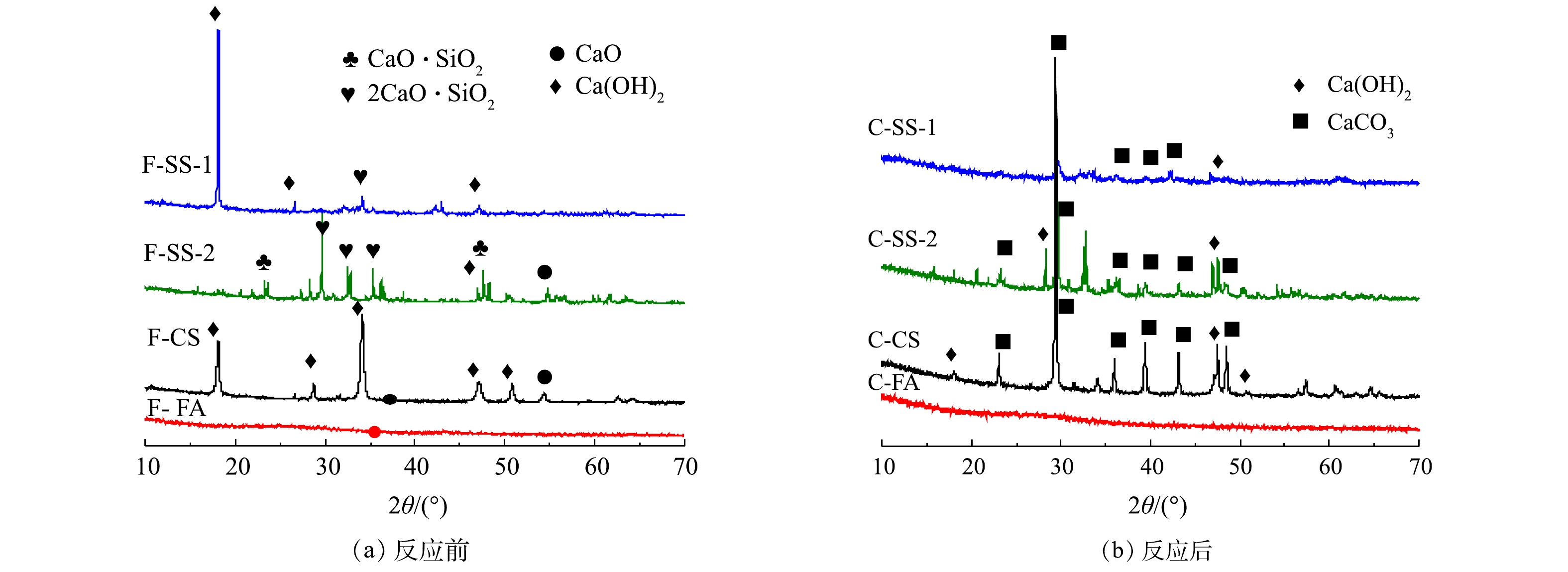
图5为碳酸化反应前后固废样品的XRD图谱。由图5可知,钢渣SS-1和SS-2原样中钙的物相存在较大差异。其中,SS-1中钙主要存在于Ca(OH)2中,而SS-2中钙主要以2CaO·SiO2和CaO·SiO2形式存在,Ca(OH)2含量较低。碳酸化反应后,SS-1中Ca(OH)2的衍射峰强度明显降低,出现较强的CaCO3的衍射峰,说明碳酸化反应中,Ca(OH)2与CO2反应生成CaCO3,且转化率较高。相比之下,SS-2反应后的样品中,出现Ca(OH)2和CaCO3的衍射峰,并存在2CaO·SiO2和CaO·SiO2的衍射峰,说明2CaO·SiO2和CaO·SiO2与CO2反应较慢。又由于SS-2钙含量比SS-1钙含量高,但SS-2的CO2封存量却比SS-1低一倍多,故可推测碳酸化封存CO2与钙基活性物相类型密切相关,2CaO·SiO2与CaO·SiO2不易发生碳酸化反应。分析其原因,可能是2CaO·SiO2和CaO·SiO2与CO2的吉布斯自由能变大于CaO和Ca(OH)2,与CO2发生反应趋势小于CaO和Ca(OH)2[25-27]。由XRD结果可知,电石渣CS样品反应前为石灰(CaO)和熟石灰(Ca(OH)2)物相,碳酸化反应后以方解石(CaCO3)为主,还有少量的石灰(CaO),而熟石灰(Ca(OH)2)的衍射峰消失,说明Ca(OH)2与CO2碳酸化反应形成CaCO3,实现CO2封存[24]。粉煤灰FA原样中未见任何含有钙组分的衍射峰,说明其中的钙为非晶相,干法反应后出现了CaCO3特征峰。这说明在干法碳酸化反应中由于非晶态钙成分不稳定,在高温环境中也能与CO2发生反应生成CaCO3,以实现CO2的封存。而由于FA中钙含量较低,FA的CO2封存量相对较低。
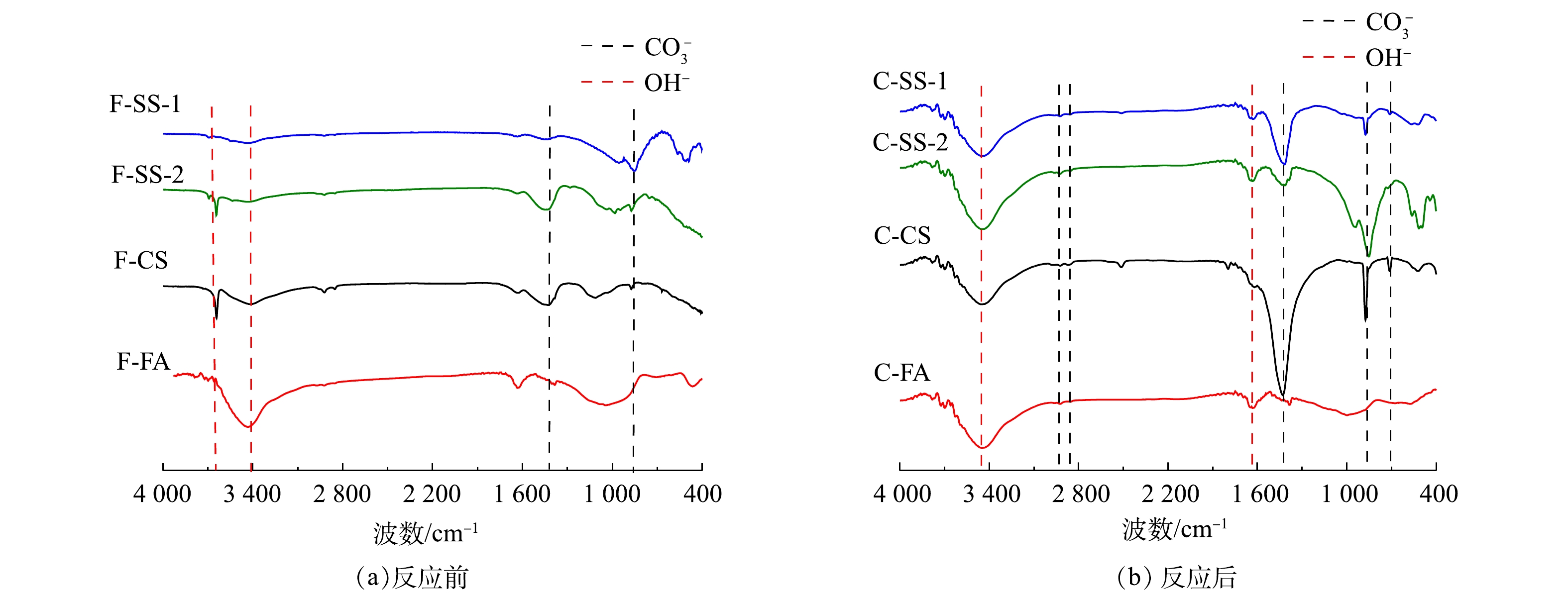
图6为碳酸化反应前后固废样品的IR谱图。反应前4个样品在3 670 cm−1、1 418 cm−1、875 cm−1和715 cm−1处均产生吸收峰,3 670 cm−1吸收峰对应于氢氧根的伸缩振动,1 418 cm−1、875 cm−1和715 cm−1吸收峰对应于碳酸盐基团的不对称伸缩振动、面外弯曲振动和面内弯曲振动[24]。上述结果表明,4个样品中均存在CaCO3。O—H可能对应于水中的氢氧键,也可能对应于Ca(OH)2中的氢氧键。由于所有样品在分析前都是烘干至恒重,不存在游离水,因此,O—H是由样品中其他状态的水分子(吸附水和结晶水)或Ca(OH)2中的—OH键伸缩振动引起的。从反应后的固废IR结果可知,SS-1、SS-2、CS样品碳酸化反应后碳酸盐基团的振动明显增强,并在2 923 cm−1和2 864 cm−1处出现了新的碳酸钙结构弱吸收峰。与XRD谱图一致,表明反应生成碳酸钙,充分证明固废样品封存了CO2。碳酸化后红外谱图中仍出现O—H的吸收谱带,表明固废中Ca(OH)2并没有完全反应,可能是被反应生成的碳酸钙所覆盖。
选取CO2封存量最大的SS-1、CS样品为例,如图7所示,反应前后样品等温吸附脱附曲线属于H3型迟滞回线,表明存在片状颗粒材料或缝形孔材料[28]。图8显示了碳酸化反应前后SS-1、CS样品比表面积及孔体积。由于灰渣在矿化过程中生成了致密的碳酸盐保护层,孔隙堵塞,造成反应后的SS-1和CS的比表面积和孔体积均小于反应前,这揭示了固废样品矿化封存CO2反应初期为快速化学反应控制阶段,反应后期为慢速扩散控制阶段的原因。
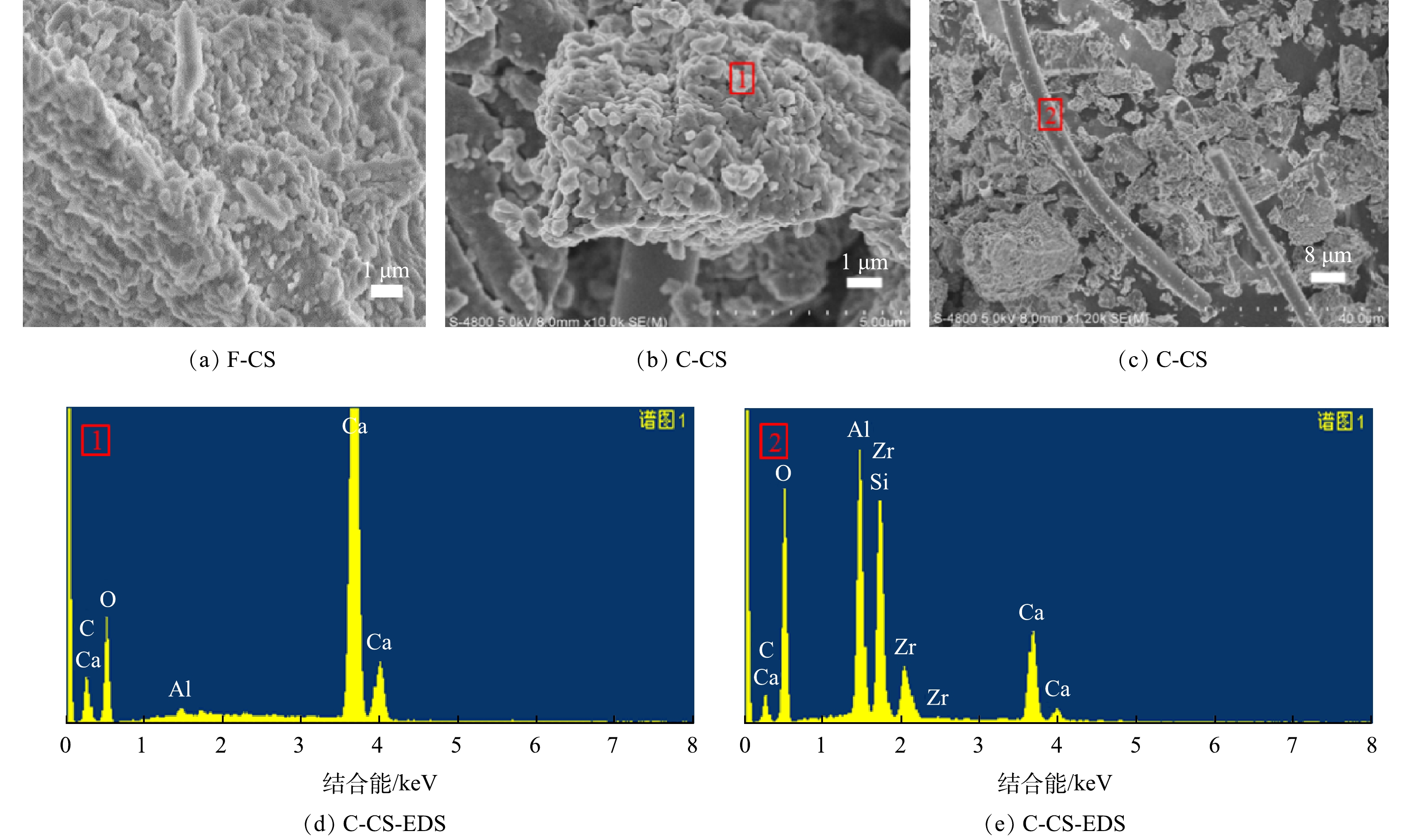
选择固废中CO2封存量最大的CS样品讨论样品碳酸化反应前后的微观形貌及微区化学组成的特征,SEM-EDS图谱见图9。CS原样形貌为无规则颗粒,碳酸化反应后外观形貌发生变化,形成了明显的块状小颗粒,堆积成块,呈现孔隙结构或缝隙结构,与等温吸附脱附曲线结果一致。结合EDS分析,证实该颗粒组成为CaCO3,与XRD、IR表征结构一致,证明样品发生碳酸化反应生成CaCO3,这也与文献[29]中得到结果一致。同时,结合EDS分析可发现,生成的微米级长柱物质组成复杂,为含有Si、Al、Ca的复合物,无规律贯穿在样品中,起到联结作用,增强样品的黏结性,从而有利于碳酸化灰渣在建材方向的资源化利用[30]。
-
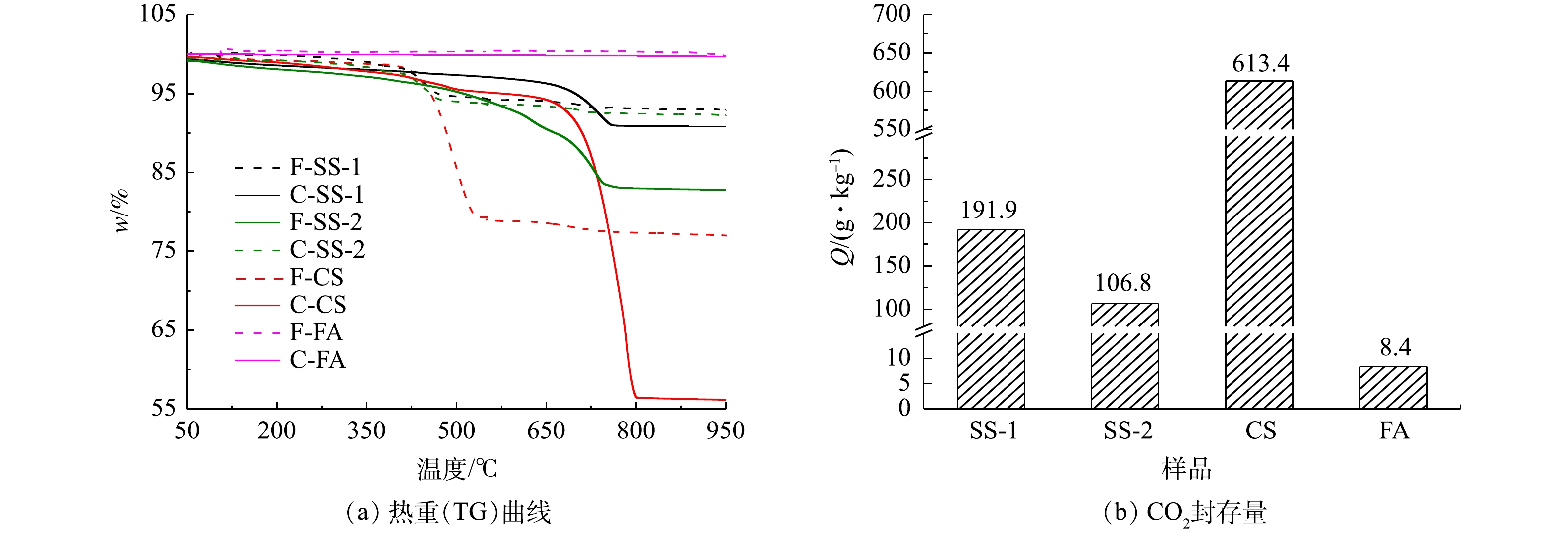
图10为样品湿法碳酸化反应前后的热重曲线。由图10可知,新鲜样品CS、SS-1、SS-2在100~540 ℃有明显质量损失。该质量损失对应Ca(OH)2分解,表明其中可能含有Ca(OH)2,这与图6中红外谱图结果一致。新鲜FA中在100~540 ℃没有出现明显质量,可认为不含Ca(OH)2。碳酸化反应后,CS,SS-1、SS-2在550~950 ℃之间出现质量损失。该质量损失对应CaCO3的分解,印证了CaCO3的生成[10]。FA反应前后变化不明显,表示其具备较低的CO2封存能力。根据计算得出,CS、SS-1、SS-2、FA湿法碳酸化封存CO2性能依次降低。
-
固废样品湿法碳酸化反应前后XRD图谱如图11所示。反应后SS-1、SS-2、CS中多为CaCO3的衍射峰,并存在少量Ca(OH)2衍射峰;而FA反应前后样品均未出现衍射峰。这表明SS-1、SS-2、CS发生碳酸化反应,形成CaCO3,但存在少量Ca(OH)2未反应。FA中的非晶相钙成分不能与CO2发生湿法碳酸化反应。此结果与样品湿法碳酸化反应前后的热重分析结果一致。
选取湿法矿化封存CO2性能最高的CS样品进行了SEM-EDS分析。如图12所示,CS原样的形貌是无规则的颗粒,湿法碳酸化反应后生成了规则的片状和菱形的立方体颗粒并聚集,或附着在原样的表面。经SEM-EDS分析,可确定该颗粒为CaCO3,表明电石渣中的CaO和Ca(OH)2会与CO2反应生成CaCO3。随着反应的进行,生成的CaCO3颗粒聚集、附着或覆盖在原样的表面,使内部的活性钙未能完全发生反应,所以反应后CS样品的XRD图谱中还存在Ca(OH)2的衍射峰。
-
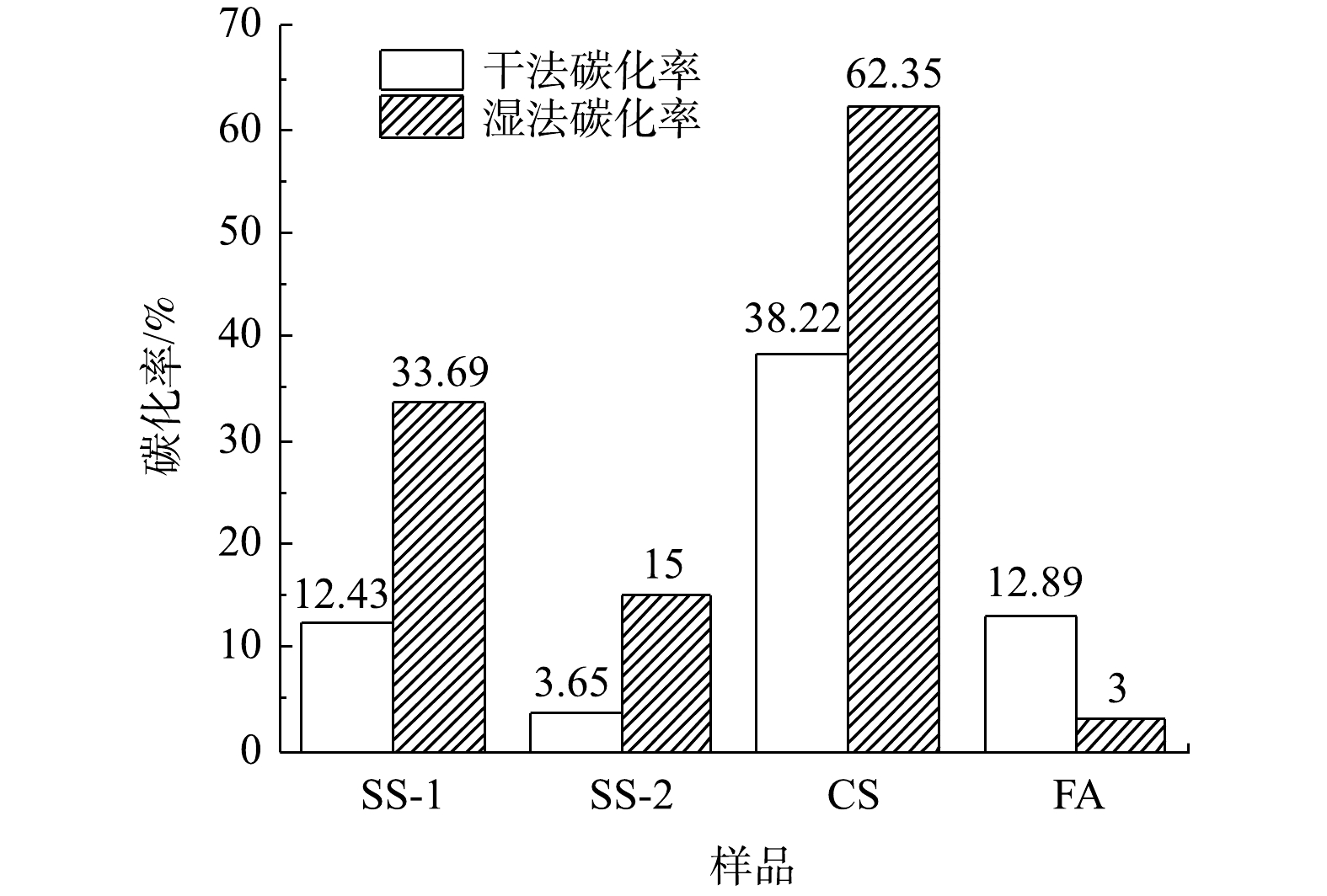
对比各种工业固废干法与湿法碳酸化CO2封存量结果(见图3和图10)可发现:干法中SS-1、SS-2、CS的CO2封存量分别为70.78、25.99和382.21 g·kg−1;湿法较干法有大幅度提升,SS-1、SS-2、CS的CO2封存量分别提升为191.9、106.8和613.4 g·kg−1。干法中FA的CO2封存量为34.81 g·kg−1,大于湿法CO2封存量的8.4 g·kg−1。而李海红[31]用电厂脱硫石膏协同粉煤灰固碳,其CO2封存量为53.108 g·kg−1,表明粉煤灰的CO2封存能力较低。这相比YANG等[32]制备CaAc2-CaO吸附剂的CO2封存量299 g·kg−1,CS、SS-1和SS-2等固废用来封存CO2更具经济优势。结合各工业固废封存能力与新鲜样品中钙组分含量,计算了干湿法固废碳化率,结果如图13所示。在干湿法矿化封存CO2工艺中,各种固废的含钙成分均未完全反应,表明其含钙组分较难发生碳酸化反应。CS样品在干湿法工艺中碳化率均为最高,湿法中最高达到62.35%,表明其中主要含钙组分易于发生碳酸化反应。由上述XRD结果可知,CS样品中Ca主要以Ca(OH)2形式存在,因此,Ca(OH)2为主要活性物种。SS-2和FA中Ca(OH)2含量最低,即活性物种最少,故其封存量及碳化率均较低。干法工艺中SS-2碳化率最低为3.65%,湿法中FA样品碳化率最低为3%。
-
通过工业固废干法与湿法碳酸化反应前后结构变化可知,碳酸化反应主要是钙基活性物种与CO2的反应。干法碳酸化过程中,CO2直接与固废中的钙基活性物种反应,接触面积小,且随着产物覆盖在固废表面阻止反应的进一步发生,会导致CO2封存量较低;而湿法碳酸化过程中水充当反应介质,促进了固废中含钙组分和CO2在水中的分散和溶解,反应更加充分。然而,并非湿法碳酸化都优于干法。由图5和图10可以看出,反应前FA中没有出现衍射峰,FA中的钙物相是以非晶相存在。干法反应后,FA出现了少量的CaCO3衍射峰,而湿法后FA依然没有出现衍射峰。这表明,干法工艺中的高温环境可使FA中不稳定的非晶相钙成分活化,与CO2接触发生反应生成CaCO3;而湿法工艺温度较低,FA中非晶相钙成分不发生反应,故呈现很低的CO2封存能力。因此,对于活性物种反应性较差的固废,如粉煤灰,其在温度较高的干法碳酸化过程中具有相对较高的碳酸化性能。
干法中碳酸化反应过程比较简单,在高温条件下,CaO等钙物相与CO2直接接触发生反应[32],反应方程式如(5)~(7)所示。
湿法中碳酸化反应过程主要包括:1)CO2溶于水生成碳酸,碳酸电离生成
$ {\rm{HCO}}_{\rm{3}}^{\rm{ - }}$ 、$ {\rm{CO}}_{\rm{3}}^{{\rm{2 - }}}$ 和H+;2)固废中的钙基活性物质溶解,在孔隙中与$ {\rm{CO}}_{\rm{3}}^{{\rm{2 - }}}$ 发生反应并生成CaCO3沉淀[33]。反应方程式如式(8)~(15)所示。从反应机理可知,湿法碳酸化过程中,CO2溶于水形成碳酸根离子后,易与固废中溶出的钙镁离子发生反应。相比之下,干法碳酸化反应中,CO2直接与含钙组分发生反应,活化能较高,尤其与CaSiO3、Ca2SiO4的反应较难进行。因此,在固废干法封存CO2过程中,即使温度远高于湿法,其封存量仍然较湿法低。另外,由反应产物(SiO2、CaCO3等)可知,反应后的固废可用于生产经济价值较低的常规建筑材料。
2.1. 不同工业固废干法矿化封存CO2性能
2.1.1. 工业固废干法矿化封存CO2效果
2.1.2. 工业固废干法矿化封存CO2速率变化
2.1.3. 工业固废干法碳酸化反应前后结构表征
2.2. 不同工业固废湿法矿化封存CO2性能
2.2.1. 工业固废湿法矿化封存CO2效果
2.2.2. 工业固废湿法碳酸化反应前后结构表征
2.3. 工业固废干法与湿法矿化封存CO2效果差异及机理对比分析
2.3.1. 工业固废干法与湿法矿化封存CO2效果差异性
2.3.2. 工业固废干法与湿法碳酸化反应机理对比分析



-
1) 3类4种大宗工业固废干法矿化封存CO2的固碳规律为CS>SS-1>FA>SS-2。湿法矿化封存CO2固碳规律为CS>SS-1>SS-2>FA。FA干法固碳能力优于湿法,CS、SS-1、SS-2湿法固碳能力优于干法。因此,可进一步开展电石渣、钢渣湿法固碳工艺和粉煤灰干法固碳工艺研发及工业化应用研究。
2) TG、XRD、IR等分析表征结果及碳酸化反应机理研究表明,钙含量是影响不同工业固废CO2封存量的关键因素,含钙硅酸盐物相是重要因素。含钙组分中,CaO和Ca(OH)2是碳酸化反应的主要活性物种,2CaO·SiO2与CaO·SiO2活性相对较差。湿法碳酸化过程中水充当反应介质,可促进固废中钙镁组分和CO2在水中的溶解,使反应更加充分。FA非晶相钙成分不稳定,在干法碳酸化反应下的高温环境能与CO2发生反应生成CaCO3,在湿法较低温度下不能发生碳酸化反应,导致FA的干法CO2封存能力高于湿法。
3)矿化封存CO2后的灰渣可用于生产经济价值较低的常规建筑材料,亦可实现工业废弃物的资源化利用。
